一种铜锡双组分复合催化剂制备方法及其应用与流程
1.本发明属于有机合成催化剂领域,特别涉及一种铜锡双组分复合催化剂制备方法及其在乙炔氢氯化反应中应用。
背景技术:
2.氯乙烯是一种非常重要的化工原料,主要用于生产聚氯乙烯树脂(pvc), pvc是世界五大工程塑料之一,在工农业生产中都具有十分广泛的应用。目前工业化的氯乙烯单体生产技术有乙炔氢氯化工艺和乙烯氧氯化工艺,我国主要采用乙炔氢氯化工艺,约占我国氯乙烯总产量的70-80%。乙炔氢氯化工艺技术简单成熟,但反应所用的氯化汞催化剂在反应过程中容易升华流失,我国必须要尽早解决氯乙烯生产中非汞催化剂的研发与替代,以稳定氯碱化工行业的可持续发展。
3.国内外学者对用于乙炔氢氯化反应的无汞催化剂的开发付出了巨大努力,对催化剂活性组分和催化剂的载体都分别进行了很多报道。最早期发现四氯化金,氯化铂等贵金属氯化物对乙炔氢氯化具有非常强的催化活性,但是贵金属催化剂价格昂贵,至今没有工业化应用报道。
4.近几年也有很多关于碳基非金属类催化剂的开发并用于乙炔氢氯化反应,比如中科院大连化物所,中科院上海高等研究院,石河子大学,天津大学,浙江大学等等,碳基非金属催化剂目前应用主要问题在于:催化剂活性相对金属催化剂要低,因此反应温度要达到200℃以上才能达到汞催化剂催化活性,但是这种非金属催化剂有个显著优点,能够耐受高反应温度,比如反应温度250℃以上可以正常使用,并且随着反应温度升高,催化活性升高。这种碳基非金属催化剂是未来无汞催化剂的一个发展方向,暂时难以直接替代氯化汞催化剂。
5.相比贵金属催化剂以及非金属催化剂的不足,非贵金属(贱金属)催化剂更具应用前景。国内外对于贱金属无汞催化剂开发,做了很多研究和报道,但是至今还是没有真正商业化。主要问题还是催化剂的活性以及稳定性不能满足工业化要求,和氯化汞催化剂催化性能还有差距。以往专利和文献中主要是围绕贱金属催化剂研究,比如铜、铋、锡等,该类催化剂需要的温度较高,但是催化剂活性低,不能满足工业化应用要求。
技术实现要素:
6.针对已经报道的非贵金属催化剂存在的问题,本发明创造一种高效稳定的非贵金属催化剂及制备方法,开发了以铜、锡为主催化剂,离子液体为助催化剂的双组分催化剂,这一催化剂在乙炔氢氯化反应中表现出了很高的活性与稳定性,相比之前报道的铜基催化剂,活性更高,活性组分用量更低,完全可以替代现行的汞触煤催化剂用于工业化生产氯乙烯,相比汞触媒这种催化剂原料广泛易得,催化剂生产,使用和后处理均不会对环境造成明显的破坏,能够完全替代现有汞催化剂使用。
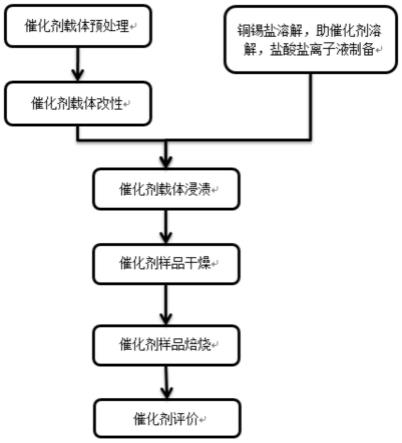
7.本发明的第一部分,本发明提供一种铜锡双组分复合催化剂制备方法,其特征在
于,包括以下步骤:
8.步骤(1):将催化剂载体,在一定条件下进行预处理,得到预处理后的催化剂载体;
9.步骤(2):将步骤(1)处理后的催化剂载体,在一定条件下使用改性剂进行改性处理,通过调控载体表面基团来提高对催化活性组分的分散和固定;
10.步骤(3):依次配制铜和锡的盐溶液;助催化剂金属盐溶液;配位离子液,将配制铜和锡的盐溶液、助催化剂金属盐溶液、配位离子液混合形成混合液;
11.步骤(4):将步骤(2)中改性后的催化剂载体与步骤(3)得到的混合液浸渍;
12.步骤(5):将浸渍后的催化剂样品进行干燥和焙烧获得用于催化乙炔氢氯化反应的铜锡双组分复合催化剂。
13.在本发明的实施例中,步骤(1)中还包括以下特征中的任一项或多项:
14.a、催化剂载体选择活性炭、生物炭、氧化铝、氧化硅、碳化硅和分子筛中的一种;
15.b、催化剂载体预处使用2n盐酸溶液或者2n硝酸溶液或者2n硫酸溶液,在30~100℃下预处理1~24小时,用去离子水洗至ph中性, 50~150℃下烘干备用。
16.进一步,优选预处理温度50~80℃,优选预处理时间5~10小时;优选干燥温度80~120℃。
17.在本发明的实施例中,步骤(2)中还包括以下特征中的任一项或多项:
18.a、对步骤(2)载体表面进行含氧,氮,硫,磷和硼原子的改性处理;含氧的前驱体主要包括醇类,酸类的化合物,包括但不限于硝酸,乙二醇,柠檬酸;含氮前驱体主要包括胺基,氨基,硝基的化合物,包括但不限于氨气,氯化铵,硝酸,丙烯酰胺,尿酸,咪唑,吡啶,吡咯,内酰胺类;含硫前驱体主要包括但不限于硫酸及硫酸盐,巯基化合物,硫脲;含磷化合物包括但不限于磷酸,磷酸盐,有机磷化合物,三苯基膦,二乙基膦;含硼前驱体包括但不限于硼酸,硼砂,有机硼化合物;
19.b、对步骤(2)中改性剂的前驱体化合物可以一种或多种用于催化剂载体改性,改性剂用量按照氧,氮,硫,磷或硼原子与载体按照质量比1:0.1~10,如1:0.1~0.3,1:0.3~0.8,1:0.8~3,1: 3~5,1:5~10;
20.c、对步骤(2)中催化剂载体的改性需要在超声波辅助下进行,超声波频率20~100khz,比如20~40khz,40~60khz,60~80khz,80~ 100khz中的一种,超声时间0.1~10小时,如0.1~2小时,2~4 小时,4~6小时,6~8小时,8~10小时;
21.d、对超声后的载体在50~150℃下干燥,如50~70℃,70~100℃, 100~120℃,120~150℃中的一种;干燥后在500~1000℃下焙烧2小时,如500~00℃,600~700℃,700~800℃,800~900℃,900~1000℃。
22.在本发明的实施例中,步骤(3)中还包括以下一项或多项:
23.a、步骤(3)中铜和锡催化剂中的催化活性组分主要包括标准电势电位高的金属盐,包含但不限于cucl2,cucl,sncl2或者sncl4中的两种或两种以上组成的铜锡双组分;活性组分总质量与载体质量比在1:(0.1~50),如1:(0.1~1),1:(1~5),1:(5-10),1:(10~20),1:(20~30), 1:(30~40),1:(40~50);
24.b、步骤(3)中助催化剂金属盐包括但不限于,licl,kcl,nacl,rbcl,cscl, bacl2,cacl2,srcl2,mgcl2,lacl3,cecl3,prcl3,eucl3;金属助剂与载体的质量比1:(0.1~100),如如1:(0.1~1),1:(1~5),1:(5-10), 1:(10~20),1:(20~30),1:(30~40),1:(40~50),
1:(50~60), 1:(60~70),1:(70~80),1:(80~90),1:(90~100)中的一种;
25.c、步骤(3)中配位离子液主要包括盐酸与亚磷酸三乙酯,亚磷酸丁酯,三苯基磷,含硫类:硫脲,甲基吡咯烷酮,n,n-二乙基乙酰胺,1,10
‑ꢀ
邻菲格林中一种或两种以上化合物形成的盐酸盐离子液体;离子液体与载体质量比1:(0.1~50),如1:(0.1~1),1:(1~5),1:(5-10), 1:(10~20),1:(20~30),1:(30~40),1:(40~50)。
26.在本发明的实施例中,步骤(4)中包括以下一项或多项:
27.a、步骤(4)中改性后的催化剂载体浸渍在步骤(3)得到的溶液中,随后进行抽真空浸渍0.1~10小时,如0.1~1小时,1~2小时,2~3小时,3~4 小时中的一种;
28.b、步骤(4)中经过抽真空浸渍后,利用超声波辅助浸渍,超声波频率 20~100khz,比如20~40khz,40-60khz,60~80khz,80~100khz中的一种,超声浸渍结束后利用旋蒸预干燥去除多余水分。
29.在本发明的实施例中,步骤(5)中包括以下一项或多项:
30.a、对浸渍后的催化剂样品在50~150℃下干燥,如50~70℃,70~ 100℃,100~120℃,120~150℃中的一种;
31.b、干燥后在500~1000℃下焙烧2小时,如500~600℃,600~700℃, 700~800℃,800~900℃,900~1000℃中的一种。
32.本发明第二部分是利用第一部分的制备方法,制备系列用于乙炔加成反应的铜锡双组分复合催化剂。
33.本发明第三部分是提供铜锡双组分复合催化剂的应用,用于乙炔氢氯化反应:主要包括把催化剂装填到固定床反应管中,通入氯化氢和乙炔进行反应。
34.优选的还包括以下一项或多项:
35.1)乙炔与氯化氢原料的摩尔比在1:(1~1.5),如1:(1~1.05),1: (1.05~1.1),1:(1.1~1.15),1:(1.15~1.2),1:(1.2~1.25), 1:(1.25~1.35),1:(1.35~1.45),1:(1.45~1.5)中的一种。
36.2)乙炔氢氯化反应体积空速,以乙炔计10~150h-1
,如10~20h-1
,20~ 40h-1
,40~60h-1
,60~80h-1
,80~90h-1
,90~100h-1
,100~120h-1
,120~ 150h-1
中的一种;
37.3)乙炔氢氯化反应压力0~1mpa,如0~0.05mpa,0.05~0.1mpa,0.1~ 0.15mpa,0.15~0.2mpa,0.2~0.3mpa,0.3~0.4mpa,0.4~0.5mpa,0.5~ 0.6mpa,0.6~0.7mpa,0.7~0.8mpa,0.8~0.9mpa,0.9~1.0mpa中的一种;
38.4)乙炔氢氯化反应温度80~180℃,如80~100℃,100~120℃,120~ 140℃,140~160℃,160~180℃中的一种。
39.本发明依据乙炔氢氯化催化反应机理,乙炔与金属盐形成的中间复合物 c2h
2-mclx越稳定,催化剂催化活性越高,产物氯乙烯选择性越高。乙炔作为供电子分子,周围电子云密度高,容易受标准电势电位高的离子或者分子吸附和活化,换言之标准电势电位高的金属离子催化乙炔氢氯化反应活性越高。依据这一原理,本发明筛选了大量标准电势电位高的非贵金属化合物作为催化剂的活性组分进行评价验证,评价结果发现标准电势电位高的金属化合物催化活性高,在此基础上获得了高活性铜锡双金属催化剂,为了进一步提高催化剂活性和稳定性,还发明了一种离子液体作为助催化剂,与双金属配位组合形成铜锡双金属复合催化剂,这种双金属双金属复合催化剂表现出了比以往文献报道的非贵金属
催化剂活性更高,稳定性更好的性能。
40.本发明的优点:
41.1)本发明采用了铜锡双组分金属离子的相互协同作用,展示了类贵金属盐特点,强力吸附活化炔烃分子,产生非常高的催化活性和催化稳定性,120 ℃,乙炔空速30h-1
,常压下,乙炔初始单程转化率可以达到95%,氯乙烯选择性大于98%。
42.2)盐酸盐离子液体作为配位化合物分子的加入提高活性组分的分散与固定,减少反应过程中损失,提高催化剂稳定性。
43.3)本发明采用非贵金属等为原料,成本远低于贵金属催化剂,并且环境友好。
附图说明
44.结合以下附图一起阅读时,将会更加充分地描述本技术内容的上述和其他特征。可以理解,这些附图仅描绘了本技术内容的若干实施方式,因此不应认为是对本技术内容范围的限定。通过采用附图,本技术内容将会得到更加明确和详细地说明。
45.图1为催化剂制备工艺流程图。
具体实施方式
46.描述以下实施例以辅助对本技术的理解,实施例不是也不应当以任何方式解释为限制本技术的保护范围。
47.具体实施方式包括两个部分:
48.第一部分一种非贵金属复合催化剂的制备的方法
49.实施例1
50.(1)称取40目的活性炭10g,用1n盐酸溶液,50℃下预处理10小时,然后用去离子水洗涤至ph中性,然后在100℃下干燥7-8小时后备用;
51.(2)将预处理后的活性炭用2n硝酸和硫酸铵,控制氮原子质量与活性炭质量比在1:8,在50khz下超声浸渍2小时,超声浸渍结束后在120℃下干燥12小时后置于管式炉中,氮气氛下焙烧2小时,得到改性后的活性炭;
52.(3)称取zncl2控制质量和活性炭比值在1:10,加水溶解得到溶液i;称取亚磷酸三乙酯和适量盐酸,控制其与活性炭质量比1:15,加水溶解得到溶液ii;
53.(4)将铜锡双组分溶液与i和ii溶液混合后,将改性活性炭加入浸渍,先抽真空浸渍2小时,然后再50khz超声波辅助下浸渍2小时,然后用旋蒸去除水分,得到初步干燥的样品;
54.(5)将初步干燥的样品在120℃下干燥5小时,然后置于500℃管式炉种氮气氛下烧2小时得到铜锡双组分复合催化剂c1;
55.实施例2
56.(1)称取40目的活性炭10g,用2n硝酸溶液,60℃下预处理3小时,然后用去离子水洗涤至ph中性,然后在120℃下干燥8小时后备用;
57.(2)将预处理后的活性炭用丙烯酰胺和硫酸铵溶液混合,控制氮和硫原子质量与活性炭质量比在1:5,在70khz下超声浸渍4小时,超声浸渍结束后在120℃下干燥8小时后置于管式炉中,氮气氛下焙烧2小时,得到改性后的活性炭;
58.(3)称取bicl3控制质量和活性炭比值在1:10,加水溶解得到溶液i;称取三苯基磷和适量盐酸混合,控制其与活性炭质量比1:20,加水溶解得到溶液ii;
59.(4)将铜锡双组分溶液与i和ii溶液混合后,将改性活性炭加入浸渍,先抽真空浸渍2小时,然后再50khz超声波辅助下浸渍2小时,然后用旋蒸去除水分,得到初步干燥的样品;
60.(5)将初步干燥的样品在120℃下干燥5小时,然后置于500℃管式炉种氮气氛下烧2小时得到铜锡双组分复合催化剂c2;
61.实施例3
62.(1)称取40目的活性炭10g,用2n盐酸溶液,60℃下预处理2小时,然后用去离子水洗涤至ph中性,然后在120℃下干燥8小时后备用;
63.(2)将预处理后的活性炭用磷酸和硫酸铵溶液混合,控制磷和硫原子质量与活性炭质量比在1:5,在70khz下超声浸渍4小时,超声浸渍结束后在 120℃下干燥8小时后置于管式炉中,氮气氛下焙烧2小时,得到改性后的活性炭;
64.(3)称取fecl3控制质量和活性炭比值在1:10,加水溶解得到溶液i;称取n-甲基吡咯烷酮和适量盐酸混合,控制其与活性炭质量比1:20,加水溶解得到溶液ii;
65.(4)将铜锡双组分溶液与i和ii溶液混合后,将改性活性炭加入浸渍,先抽真空浸渍2小时,然后再50khz超声波辅助下浸渍2小时,然后用旋蒸去除水分,得到初步干燥的样品;
66.(5)将初步干燥的样品在120℃下干燥5小时,然后置于500℃管式炉种氮气氛下烧2小时得到铜锡双组分复合催化剂c3;
67.实施例4
68.(1)称取40目的活性炭10g,用1n盐酸溶液,50℃下预处理10小时,然后用去离子水洗涤至ph中性,然后在100℃下干燥7-8小时后备用;
69.(2)将预处理后的活性炭用2n硼酸和硫酸铵,控制硼和氮原子质量与活性炭质量比在1:8,在50khz下超声浸渍2小时,超声浸渍结束后在120 ℃下干燥12小时后置于管式炉中,氮气氛下焙烧2小时,得到改性后的活性炭;
70.(3)称取srcl2控制质量和活性炭比值在1:10,加水溶解得到溶液i;称取吡咯烷酮和适量盐酸,控制其与活性炭质量比1:15,加水溶解得到溶液 ii;
71.(4)将铜锡双组分溶液与i和ii溶液混合后,将改性活性炭加入浸渍,先抽真空浸渍2小时,然后再50khz超声波辅助下浸渍2小时,然后用旋蒸去除水分,得到初步干燥的样品;
72.(5)将初步干燥的样品在120℃下干燥5小时,然后置于500℃管式炉种氮气氛下烧2小时得到铜锡双组分复合催化剂c4;
73.实施例5
74.(1)称取50目的活性炭10g,用2n盐酸溶液,60℃下预处理10小时,然后用去离子水洗涤至ph中性,然后在130℃下干燥5小时后备用;
75.(2)将预处理后的活性炭用磷酸氢氨和硼砂溶液混合,控制磷和硼原子质量与活性炭质量比在1:10,在60khz下超声浸渍4小时,超声浸渍结束后在150℃下干燥4小时后置于管式炉中,氮气氛下焙烧2小时,得到改性后的活性炭;
76.(3)称取cecl2控制质量和活性炭比值在1:10,加水溶解得到溶液i;称取己内酰胺和适量盐酸,控制其与活性炭质量比1:15,加水溶解得到溶液 ii;
77.(4)将铜锡双组分溶液与i和ii溶液混合后,将改性活性炭加入浸渍,先抽真空浸渍2小时,然后再50khz超声波辅助下浸渍2小时,然后用旋蒸去除水分,得到初步干燥的样品;
78.(5)将初步干燥的样品在120℃下干燥5小时,然后置于500℃管式炉种氮气氛下烧2小时得到铜锡双组分复合催化剂c5;
79.对比例1
80.(1)称取40目的活性炭10g,用2n盐酸溶液,60℃下预处理6小时,然后用去离子水洗涤至ph中性,然后在120℃下干燥5小时后备用;
81.(2)将预处理后的活性炭用吡啶和硼砂溶液混合,控制氮和硼原子质量与活性炭质量比在1:15,在60khz下超声浸渍8小时,超声浸渍结束后在60 ℃下干燥16小时后置于管式炉中,氮气氛下焙烧2小时,得到改性后的活性炭;
82.(3)称取cucl2,控制质量和活性炭比值在1:10,加水溶解得到溶液i;
83.(4)将改性活性炭加入浸渍溶液i中,先抽真空浸渍5小时,然后再70khz 超声波辅助下浸渍4小时,然后用旋蒸去除水分,得到初步干燥的样品;
84.(5)将初步干燥的样品在120℃下干燥6小时,然后置于500℃管式炉种氮气氛下烧2小时得到非汞催化剂c6;
85.对比例2
86.(1)称取40目的活性炭10g,用2n盐酸溶液,60℃下预处理6小时,然后用去离子水洗涤至ph中性,然后在120℃下干燥5小时后备用;
87.(2)将预处理后的活性炭用吡啶和硼砂溶液混合,控制氮和硼原子质量与活性炭质量比在1:10,在60khz下超声浸渍6小时,超声浸渍结束后在60 ℃下干燥12小时后置于管式炉中,氮气氛下焙烧2小时,得到改性后的活性炭;
88.(3)称取sncl2控制质量和活性炭比值在1:10,加水溶解得到溶液i;
89.(4)将改性活性炭加入溶液i中浸渍,先抽真空浸渍4小时,然后再60khz 超声波辅助下浸渍5小时,然后用旋蒸去除水分,得到初步干燥的样品;
90.(5)将初步干燥的样品在130℃下干燥8小时,然后置于500℃管式炉种氮气氛下烧2小时得到非汞催化剂c7;
91.第二部分将第一部分制备的c1-c7催化剂用于反应中评价。
92.1.催化乙炔氢氯化反应:
93.取c1-c7催化剂样品1g,装入直径10mm的反应管中,固定床反应器评价催化剂活性和稳定性,乙炔和氯化氢摩尔比1:(1~1.5),乙炔空速10~150h-1
,反应温度80~180℃,反应压力0~1mpa,气相产物用气相色谱分析。催化剂评价结果见表1(表中乙炔转化率为初始最高转化率)。
94.表1c1-c7催化剂评价结果
[0095][0096]
尽管本技术已公开了多个方面和实施方式,但是其它方面和实施方式对本领域技术人员而言将是显而易见的,在不脱离本技术构思的前提下,还可以做出若干变形和改进,这些都属于本技术的保护范围。本技术公开的多个方面和实施方式仅用于举例说明,其并非旨在限制本技术,本技术的实际保护范围以权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1