一种N、S共掺杂碳材料负载锌单原子及其制备方法和应用与流程
一种n、s共掺杂碳材料负载锌单原子及其制备方法和应用
技术领域
1.本发明涉及有机合成以及单原子催化剂技术领域,具体涉及一种n、s共掺杂碳材料负载锌单原子及其制备方法和应用。
背景技术:
2.胺类化合物是化工领域的中心化合物,在农用化学品、医药、催化配体、染料和生物活性化合物方面有着广泛的应用。以醇为烷基化试剂,通过借氢策略与胺反应,只生成水作为副产物,为胺类化合物的制备提供了绿色环保的方法。
3.在此背景下,ru、ir、pd等催化体系首先应用(acs catal6 2613,3,6,565-511)(greenchem62617,19,3142-3161)(journal of colloid and interface science 2622,666,1346-1361),随后 fe、co、mn、mo、w等金属的借氢能力也被发掘(org6 lett6 2616,17,1656-1659)(org6 chem. front6,2619,6,562-567)(org6 lett6 2619,21,3142-3147)(acs catal6 2621,11,16377-16352)。然而,这些方法都存在一些缺点,例如原子利用率低;使用ru、ir、pd等贵金属催化剂价格昂贵,无法大规模使用,或反应体系中往往需要加入1-3当量的碱,反应条件苛刻;均相催化体系不利于产物分离且无法对催化剂进行回收利用。
4.近年来,锌催化体系构建c-c、c-n键进入研究者视野。2626年,mannathan等人报道了六水合硝酸锌,催化醇和胺的n-烷基化反应,首次证明了zn在n-烷基化反应催化能力(adv6 synth6catal62626,362,4469
–
44)。然而,该催化体系条件较苛刻(146℃,36h),且均相催化剂无法回收再利用。
技术实现要素:
5.本发明的目的是克服现有技术的一个或多个不足,提供一种新型的n、s共掺杂碳材料负载锌单原子的制备方法,该方法制成的n、s共掺杂碳材料负载锌单原子可作为催化剂高效、低成本且反应条件更温和地合成胺类化合物,同时稳定性高,可回收。
6.本发明同时还提供了一种上述方法制成的n、s共掺杂碳材料负载锌单原子。
7.本发明同时还提供了一种上述方法制成的n、s共掺杂碳材料负载锌单原子在催化胺和醇的借氢胺化反应中的应用。
8.为达到上述目的,本发明采用的一种技术方案是:
9.一种n、s共掺杂碳材料负载锌单原子的制备方法,该制备方法包括:
10.将2-甲基咪唑、苯胺、氨水和硫脲混合,分散于溶剂中,获得第一混合物;
11.将硝酸锌水溶液与所述第一混合物混合,球磨分散,得到第二混合物,洗涤,干燥,获得前驱体;
12.将该前驱体在惰性气氛下煅烧,得到所述n、s共掺杂碳材料负载锌单原子;
13.其中,控制所述2-甲基咪唑、所述苯胺、所述硫脲和所述硝酸锌的投料摩尔量比为 2-6∶2-6∶666-166∶1,所述氨水中的氨与所述硝酸锌的投料摩尔量比为266-12∶1。
14.根据本发明的一些优选且具体的方面,控制所述2-甲基咪唑、所述苯胺、所述硫脲和所述硝酸锌的投料摩尔量比为3-6∶3-6∶665-162∶1。
15.根据本发明的一个具体方面,所述氨水可以选用分析纯试剂,浓度约为26wt6%-25wt6%,所述氨水的用量相对于所述硝酸锌的用量为:662-665ml/mmol。
16.根据本发明的一些优选方面,将2-甲基咪唑、苯胺、氨水和硫脲混合并分散于溶剂之前,先将2-甲基咪唑、苯胺、氨水和硫脲混合并进行球磨。在本发明的一些实施方式中,在球磨过程中,可以采用氧化锆作为磨球,便于获得均一的混合物。
17.根据本发明的一些优选方面,所述溶剂为选自可以溶解各成分的有机溶剂,具体可以为选自甲醇、乙醇和丙醇中的一种或多种的组合。
18.根据本发明的一些优选方面,所述洗涤采用去离子水离心洗涤,所述干燥采用真空干燥的方式进行。在本发明的一些实施方式中,真空干燥在76-96℃下进行。
19.根据本发明的一些优选方面,所述惰性气氛为氮气气氛和/或氩气气氛。
20.根据本发明的一些具体方面,可以采用将六水合硝酸锌溶解于去离子水中得到硝酸锌水溶液。
21.根据本发明的一些具体方面,得到第二混合物之前的球磨分散可以采用如下方式进行:转速为366-666rpm,球磨1-6小时;其中,每转动26-46min,暂停2-16min,重复该过程,直至球磨结束。
22.根据本发明的一些优选方面,所述煅烧的煅烧温度为566-1666℃。进一步地,所述煅烧的煅烧温度为566-966℃。
23.根据本发明的一些优选方面,所述煅烧的煅烧时间为1-6h。进一步地,所述煅烧的煅烧时间为1-6h。
24.根据本发明的一些优选方面,控制从室温升温至所述煅烧温度的升温速率为3-5℃/min。
25.本发明提供的又一技术方案:一种上述所述的制备方法制成的n、s共掺杂碳材料负载锌单原子。
26.根据本发明,以重量百分含量计,该n、s共掺杂碳材料负载锌单原子中锌的负载量为2 wt6%-6wt6%。进一步地,以重量百分含量计,该n、s共掺杂碳材料负载锌单原子中锌的负载量为266wt6%-466wt6%。
27.本发明提供的又一技术方案:一种上述所述的n、s共掺杂碳材料负载锌单原子作为催化剂在芳香胺与醇的借氢n-烷基化反应中的应用。
28.根据本发明的一些优选且具体的方面,使芳香胺与醇在碱存在下、在溶剂中、在惰性气氛下、在116-136℃下以及在n、s共掺杂碳材料负载锌单原子催化下反应生成对应胺类化合物。
29.根据本发明的一些优选方面,在借氢n-烷基化反应中,所述反应的反应温度为116-126℃。
30.根据本发明,在借氢n-烷基化反应中,所述反应的时间为6-26h。
31.根据本发明,所述n、s共掺杂碳材料负载锌单原子在反应结束后进行回收再利用,重复利用的次数可以达到5次及以上,且催化活性无明显衰减。
32.根据本发明的一些优选方面,在借氢n-烷基化反应中,溶剂可以为甲苯、乙苯等。
33.根据本发明的一些优选方面,在借氢n-烷基化反应中,碱可以为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾等。
34.根据本发明的一些优选方面,在借氢n-烷基化反应中,芳香胺、醇、碱、锌的用量摩尔比为1:166-266:661-666:66661-661。进一步地,借氢n-烷基化反应中,芳香胺、醇、碱、锌的用量摩尔比为1:165-262:6616-6646:66663-6666。
35.根据本发明的一个具体方面,在借氢n-烷基化反应中,所述n、s共掺杂碳材料负载锌单原子与所述芳香胺的用量比为16-26mg/mmol。
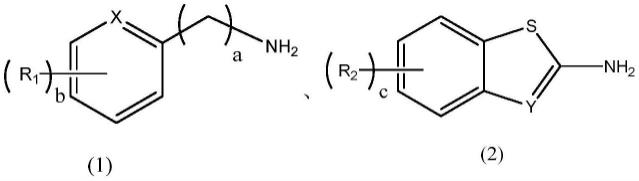
36.根据本发明的一些优选且具体的方面,在借氢n-烷基化反应中,芳香胺可以为选自如下结构中的一种或多种:
[0037][0038]
式(1)中,r1为选自h、c
1-6
烷基、卤素、芳香基或c
1-6
烷氧基,a可以为6、1、2、3、 4或6,b可以为1、2、3、4、6、6、7、5、9或16,x为ch或n;
[0039]
式(2)中,r2为选自h、c
1-6
烷基、卤素、芳香基或c
1-6
烷氧基,c可以为1、2、3或 4,y为ch或n。
[0040]
进一步地,芳香胺可以为
[0041]
进一步优选地,c
1-6
烷基可以为取代或未取代的如下基团:甲基、乙基、丙基、异丙基、正丁基、异丁基、正戊基、异戊基、己基等;卤素可以为氟、氯或溴;芳香基可以为取代或未取代的苯基;c
1-6
烷氧基可以为取代或未取代的如下基团:甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、异丁氧基等。
[0042]
根据本发明,在借氢n-烷基化反应中,醇不做具体限制,可以为脂肪醇、芳香醇等,例如可以为取代或未被取代的c
1-16
烷基醇、取代或未被取代的α-苯基烷基醇、羟基取代的杂环、含有烯醇的化合物等。其中,杂环可以为3-16元杂环,杂环的杂原子可以为氧、硫和氮中的一个或多个。
[0043]
在本发明的一些实施方式中,醇可以为甲醇、乙醇、丙醇、正丁醇、正戊醇、正己醇、正庚醇、正辛醇、异戊醇、异丙醇、异辛醇、
等;其中,a为ch或n,b为s或o,m为ch或n,d、i独立地为1、2、3、4、6、6、7、5、9或16,e、g独立地为6、1、2、3、4、6或6。
[0044]
由于上述技术方案运用,本发明与现有技术相比具有下列优点:
[0045]
本发明经过不断地实验,研究发现,通过采用上述特定的化合物组合进行n、s的掺杂,有效地改善了zn中心的电子结构,使反应可以较为温和且短时间完成;独特的原位掺杂碳化方式,增加了催化剂的比表面积和孔体积,有利于对反应物的吸附,增加催化效率;该n、s 共掺杂碳材料负载锌单原子催化剂对胺和醇的借氢n-烷基化反应表现出优异的催化性能,产率高且经济环保,避免了贵金属催化剂的使用;同时底物适用范围广,对于直链醇也可以反应,且反应结束后催化剂可以回收再利用。
附图说明
[0046]
图1为本发明实施例1制备的n、s共掺杂碳材料负载锌单原子前驱体的xrd图。
[0047]
图2为本发明实施例1制备的n、s共掺杂碳材料负载锌单原子前驱体的sem图。
[0048]
图3为本发明实施例1制备的n、s共掺杂碳材料负载锌单原子的xrd图。
[0049]
图4为本发明实施例1制备的n、s共掺杂碳材料负载锌单原子的tem图。
[0050]
图5为本发明实施例1制备的n、s共掺杂碳材料负载锌单原子的 haadf-stem-eds图;
[0051]
图6为本发明实施例1制备的n、s共掺杂碳材料负载锌单原子的xanes和exafs图。
具体实施方式
[0052]
以下结合具体实施例对上述方案做进一步说明;应理解,这些实施例是用于说明本发明的基本原理、主要特征和优点,而本发明不受以下实施例的范围限制;实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。
[0053]
下述实施例中未作特殊说明,所有原料均来自于商购或通过本领域的常规方法制备而得。
[0054]
实施例1
[0055]
本例提供一种n、s共掺杂碳材料负载锌单原子的制备方法,该方法包括如下步骤:
[0056]
步骤1,将2-甲基咪唑(24mmol)、苯胺(24mmol)、氨水(3ml,市售分析纯氨水)和硫脲 (6mmol)加入到氧化锆球磨罐中,用乙醇(16ml)溶解获得均一溶液a。
[0057]
步骤2,将六水合硝酸锌(6mmol)溶于去离子水(6ml)中,获得硝酸锌水溶液b。
[0058]
步骤3,将b液倒入a液中,在466rpm转速下,球磨三个小时,得到粘稠白色液体。样品用去离子水离心洗涤,56℃真空干燥,获得前驱体zif-5-aa@s(其xrd图和sem图分别如图1和图2所示)。
[0059]
步骤4,将前驱体在管式炉中以6℃/min的升温速率,966℃、氮气气氛下,煅烧2h,制得所述n、s共掺杂碳材料负载锌单原子,简称cns@zn
1-aa。其中锌含量为3676wt6%(采用icp测试)。
[0060]
其xrd图、tem图、haadf-stem-eds图、xanes和exafs图分别如图3、图4、图6和图6所示,其中:
[0061]
图1:根据zif-5-aa@s的xrd图谱结果表明,制备的前驱体材料具有完整的zif-5结构。
[0062]
图2:根据zif-5-aa@s的sem图表明,前驱体为约266nm尺寸的微球型。
[0063]
图3:在cns@zn
1-aa的xrd图谱中,在24
°
和43
°
处有两个宽峰,它们分别归属于石墨碳材料的(662)和(161)晶面。没有检测到锌物质,不包括大晶体锌颗粒的存在。只检测出石墨碳的衍射峰,没有观察到zn原子的峰,表明锌在该催化剂中可能以单原子形式存在。
[0064]
图4:在tem中没有观察到高度结晶的zn物质,这与xrd的结果一致。
[0065]
图6:haadf-stem-eds图中,用圆圈标记的大量亮点代表孤立的锌原子。edx元素映射图像显示均匀分布的n、o、s和zn信号,这表明zn、n和s已成功掺杂到碳基质中。
[0066]
图6:根据xanes和exafs结果,锌在该材料中以单原子形式存在,第一壳层外存在 3~4个氮,1个硫。
[0067]
实施例2
[0068]
制备催化剂的步骤基本同实施例1,区别在于实施例1所述的制备方法中,苯胺用量由 24mmol调整为21mmol,制得n、s共掺杂碳材料负载锌单原子。
[0069]
实施例3
[0070]
制备催化剂的步骤基本同实施例1,区别在于实施例1所述的制备方法中,氨水用量由3ml调整为2.5ml,制得所述n、s共掺杂碳材料负载锌单原子。
[0071]
实施例4
[0072]
本实施例提供芳香胺与醇的借氢n-烷基化反应制备过程,具体地:
[0073]
将1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg实施例1制备的 cns@zn
1-aa置于26ml密封管中,氮气保护。加热到126℃,反应12h。反应完成后,密封管冷却至室温,离心分离催化剂及溶液,溶液用气相色谱法(gc)和气相色谱质谱法(gc-ms) 分析,3a产率为96%。
[0074]
该反应过程示意如下:
[0075]
对比例1
[0076]
制备催化剂的步骤基本同实施例1,区别在于实施例1所述的制备方法中,不添加苯胺,前驱体名称为zif-5-am@s,煅烧获得cns@zn
1-am。
[0077]
将1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg cns@zn
1-am置于26ml 密封管中,氮气保护。加热到126℃,反应12h。反应完成后,密封管冷却至室温,离心分离催化剂及溶液。溶液用气相色谱法(gc)和气相色谱质谱法(gc-ms)分析,3a产率为6%。
[0078]
对比例2
[0079]
制备催化剂的步骤基本同实施例1,区别在于实施例1所述的制备方法中,不添加硫脲,前驱体名称为zif-5,煅烧获得cn@zn1。
[0080]
将1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg cn@zn1置于26ml密封管中,氮气保护。加热到126℃,反应12h。反应完成后,密封管冷却至室温,离心分离催化剂
及溶液。溶液用气相色谱法(gc)和气相色谱质谱法(gc-ms)分析,3a产率为34%。
[0081]
对比例3
[0082]
制备催化剂的步骤基本同实施例1,区别在于实施例1所述的制备方法中,用等摩尔的三苯基膦代替硫脲,前驱体名称为zif-5@p,煅烧获得cnp@zn1。
[0083]
将1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg cnp@zn1置于26ml 密封管中,氮气保护。加热到126℃,反应12h。反应完成后,密封管冷却至室温,离心分离催化剂及溶液。溶液用气相色谱法(gc)和气相色谱质谱法(gc-ms)分析,3a产率为62%。
[0084]
对比例4
[0085]
制备催化剂的步骤基本同实施例1,区别在于实施例1所述的制备方法中,不添加氨水,前驱体名称为zif-5-an@s,煅烧获得cns-2@zn
1-an。
[0086]
将1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg cns@zn
1-an置于26ml 密封管中,氮气保护。加热到126℃,反应12h。反应完成后,密封管冷却至室温,离心分离催化剂及溶液。溶液用气相色谱法(gc)和气相色谱质谱法(gc-ms)分析,3a产率为6%。
[0087]
对比例5
[0088]
制备催化剂的步骤基本同实施例1,区别在于实施例1所述的制备方法中,将2-甲基咪唑的用量由24mmol调整为4mmol,前驱体名称为zif-5-aa-2@s,煅烧获得cns-2@zn
1-aa。
[0089]
将1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg cns-2@zn
1-aa 置于26ml密封管中,氮气保护。加热到126℃,反应12h。反应完成后,密封管冷却至室温,离心分离催化剂及溶液。溶液用气相色谱法(gc)和气相色谱质谱法(gc-ms)分析,3a产率为 16%。
[0090]
实施例5-18
[0091]
本些实施例提供芳香胺与醇的借氢n-烷基化反应制备过程,基本同实施例4,仅改变原料芳香胺和醇中的一个或2个,具体结果参见表1所示。
[0092]
表1
[0093]
[0094][0095]
表中,-ph为苯基,-ome为甲氧基。
[0096]
实施例19:
[0097]
本实施例提供芳香胺与醇的借氢n-烷基化反应制备过程同实施例4,其区别仅在于:催化剂采用实施例2制备的n、s共掺杂碳材料负载锌单原子,3a产率为55%。
[0098]
实施例20:
[0099]
本实施例提供芳香胺与醇的借氢n-烷基化反应制备过程同实施例4,其区别仅在于:催化剂采用实施例3制备的n、s共掺杂碳材料负载锌单原子,3a产率为59%。
[0100]
实施例21:催化剂重复使用活性测试
[0101][0102]
将1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg实施例1制备的 cns@zn
1-aa置于26ml密封管中,氮气保护。加热到126℃,反应12h。反应完成后,密封管冷却至室温,离心,分离催化剂及溶液。催化剂用少量水和乙酸乙酯洗涤后转入26ml密封管中,加入1mmol苯胺、2mmol苯甲醇、663mmol koh、2ml甲苯和16mg cns@zn
1-aa,氮气保护,加热到126℃,反应12h。反应结束,密封管冷却至室温,重复催化剂回收及套用投料操作。经气相色谱法(gc)和气相色谱质谱法(gc-ms)分析,目标产物3a在催化剂新用和套用1-5次的反应中的产率分别如下:96%,96%,59%,96%,59%,96%,59%,59%,59%。催化剂活性无明显衰减。
[0103]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
[0104]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1