一种饲料中真菌毒素的提取方法和检测方法与流程
文档序号:31775715发布日期:2022-10-12 08:23阅读:1034来源:国知局
导航: X技术> 最新专利>物理化学装置的制造及其应用技术
1.本发明涉及分析检测技术领域,具体涉及一种饲料中真菌毒素的提取方法和检测方法。
背景技术:
2.链格孢属真菌属于丝状真菌,是一种普遍存在于水果、蔬菜、田间作物以及存储的饲料等中的病原体和腐生菌,由于它们可以在低温、潮湿的环境下生长繁殖,因此是导致冷藏或长途运输过程中水果、蔬菜、谷物、饲料等腐烂变质的重要病原菌之一,产生的毒素不仅严重危害人类健康和畜禽生产安全,也造成了经济损失。
3.链格孢霉毒素主要包括交链孢酚(alternariol,aoh)、交链孢烯(altenuene,alt)、交链孢毒素ii(altertoxins ii,atx ii)和腾毒素(tentoxin,ten)等。目前,我国已制定饲料中黄曲霉毒素b1,赭曲霉毒素a、玉米赤霉烯酮、呕吐毒素、t-2毒素和伏马毒素的限量标准,但缺乏链格孢霉毒素的限量标准。链格孢霉毒素现用的检测方法有薄层色谱分析、液相色谱法、气相色谱、气相色谱与质谱联用仪,超高效液相色谱-串联质谱法和竞争酶联免疫吸附法等,但是这些方法适用基质不包含饲料,而且检测种类较少。因此,有必要建立饲料中链格孢霉毒素的快速、灵敏分析新技术。
技术实现要素:
4.本发明的目的在于提供一种饲料中真菌毒素的提取方法和检测方法,采用本发明提供的方法能够快速、准确的实现饲料中交链孢酚、交链孢烯、玉米赤霉烯酮、交链孢毒素ii和腾毒素的定性与定量检测。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种饲料中真菌毒素的提取方法,包括以下步骤:
7.将待测饲料与乙腈混合进行超声提取,将得到提取物溶解于甲醇中,得到甲醇复溶液;
8.将所述甲醇复溶液依次进行吸附净化和正己烷洗涤,得到真菌毒素提取液;所述吸附净化用吸附剂包括c18吸附剂和psa吸附剂;
9.所述真菌毒素包括交链孢酚、交链孢烯、玉米赤霉烯酮、交链孢毒素ii和腾毒素中的一种或几种。
10.优选的,所述待测饲料的质量与乙腈的体积之比为1g:5~10ml。
11.优选的,所述待测饲料包括渔用饲料。
12.优选的,所述超声提取的功率≥600w,温度为40~45℃,时间≥30min。
13.优选的,所述待测饲料的质量与甲醇的体积之比为1g:0.5~1ml。
14.优选的,所述待测饲料和吸附剂的质量比为1:0.2~0.6。
15.优选的,所述c18吸附剂的粒径为40~50μm。
16.优选的,所述psa吸附剂为乙二胺-n-丙基硅烷化硅胶;所述psa吸附剂的粒径为40
~60μm。
17.本发明提供了一种饲料中真菌毒素的检测方法,包括以下步骤:
18.对上述技术方案所述提取方法得到的真菌毒素提取液中的真菌毒素进行液相色谱串联质谱检测。
19.优选的,所述液相色谱的检测条件包括:色谱柱为c18柱;柱箱温度为40℃;进样量为5μl;流动相体系为流动相a和流动相b,所述流动相a为甲醇,所述流动相b为水;所述流动相体系的流速为0.3ml/min;洗脱方式为梯度洗脱;
20.所述梯度洗脱的程序为:
21.0.00~2.00min:所述流动相a的体积百分含量为90%;
22.2.00~4.00min:所述流动相a的体积百分含量由10%匀速增加到95%;
23.4.00~8.00min:所述流动相a的体积百分含量为95%;
24.8.00~8.10min:所述流动相a的体积百分含量由95%匀速减少到10%;
25.8.10~10.00min:所述流动相a的体积百分含量为10%;
26.所述质谱的检测条件包括:离子源为电喷雾离子源;检测方式为多反应监测;扫描方式为负离子模式;喷雾电压为3.0kv;离子传输管温度为320℃;脱溶剂气温度为300℃。
27.本发明提供了一种真菌毒素的提取方法,包括以下步骤:将待测饲料与乙腈混合进行超声提取,将得到提取物溶解于甲醇中,得到甲醇复溶液;将所述甲醇复溶液依次进行吸附净化和正己烷洗涤,得到真菌毒素提取液;所述吸附净化用吸附剂包括c18吸附剂和psa吸附剂;所述真菌毒素包括交链孢酚、交链孢烯、玉米赤霉烯酮、交链孢毒素ii和腾毒素中的一种或几种。本发明提供的检测方法,采用乙腈作为提取溶剂,对饲料中交链孢酚、交链孢烯、玉米赤霉烯酮、交链孢毒素ii和腾毒素的提取率高,进而提高检测的灵敏度、准确性和稳定性;而且,与振荡提取和均质提取相比,本发明采用超声提取能够实现多个样品的高通量处理,处理效率高,检测效率高。通过c18吸附剂和psa吸附剂进行净化,能够很好的除去饲料中的大部分色素、脂类、蛋白质、有机酸等杂质,通过正己烷净化能够进一步去除脂类等弱极性杂质,对5种真菌毒素净化效果好,进一步提高了后续液相色谱串联质谱法对5种真菌毒素检测的灵敏度、准确性和稳定性。
28.本发明还提供了一种饲料中真菌毒素的检测方法,包括以下步骤:对上述技术方案所述提取方法得到的真菌毒素提取液进行液相色谱串联质谱检测。采用本发明提供的提取方法得到的真菌毒素提取液杂质少,液相色谱串联质谱法对5种真菌毒素检测的灵敏度、准确性和稳定性,采用液相色谱串联质谱法实现了5种真菌毒素的定性和定量检测,为饲料检验提供技术支撑,弥补了目前针对交链孢酚、交链孢烯、交链孢毒素ii和腾毒素4种物质检测方法的空白。
29.如实施例测试结果所示,本发明提供的检测方法的平均加标回收率75.1%~87.3%(准确度,n=6,满足60%~120%要求),相对标准偏差(变异系数,rsd)3.33%~9.05%。说明,本发明提供方法的提取和净化效果好,灵敏度高、准确性和稳定性好,适合于饲料样品中真菌毒素的多残留、高通量检测。
附图说明
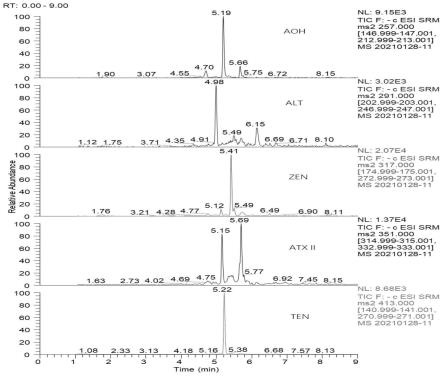
30.图1为加标样品溶液mrm谱图;
31.图2为不同提取剂下真菌毒素的回收率结果图;
32.图3为振荡提取采用的振荡器图;
33.图4为均质提取采用的均质器图;
34.图5为不同提取方式下真菌毒素的回收率结果图;
35.图6为c18吸附剂和正己烷联合净化的mrm谱图;
36.图7为psa吸附剂和正己烷联合净化的mrm谱图;
37.图8为c18吸附剂、psa吸附剂和正己烷联合净化的mrm谱图;
38.图9为乙腈和0.1%(v/v)甲酸水溶液作为流动相时的mrm谱图;
39.图10为乙腈和0.1%(v/v)甲酸-乙酸铵水溶液(乙酸铵浓度为5mmol/l)作为流动相时的mrm谱图
‘
40.图11为乙腈和水作为流动相时的mrm谱图;
41.图12为甲醇和0.1%(v/v)甲酸-乙酸铵水溶液(乙酸铵浓度为5mmol/l)作为流动相时的mrm谱图;
42.图13为甲醇和0.1%(v/v)甲酸水溶液作为流动相时的mrm谱图;
43.图14为甲醇和水作为流动相时的标准溶液mrm谱图。
具体实施方式
44.本发明提供了一种饲料中真菌毒素的提取方法,包括以下步骤:
45.将待测饲料与乙腈混合进行超声提取,将得到提取物溶解于甲醇中,得到甲醇复溶液;
46.将所述甲醇复溶液依次进行吸附净化和正己烷洗涤,得到真菌毒素提取液;所述吸附净化用吸附剂包括c18吸附剂和psa吸附剂;
47.所述真菌毒素包括交链孢酚、交链孢烯、玉米赤霉烯酮、交链孢毒素ii和腾毒素中的一种或几种。
48.在本发明中,若没有特殊说明,所采用的试剂均为本领域技术人员所熟知的市售商品。
49.本发明将待测饲料与乙腈混合进行超声提取,将得到提取物溶解于甲醇中,得到甲醇复溶液。
50.在本发明中,所述待测饲料优选包括渔用饲料,更优选包括鱼饲料、虾饲料、蟹饲料和海参饲料中的一种或几种。在本发明中,所述待测饲料在使用前优选先依次进行粉碎或均质和过筛,本发明对于所述粉碎没有特殊限定,采用本领域技术人员熟知的粉碎方式即可。在本发明中,所述均质优选利用破壁机或均质器进行,所述均质的速度优选为20000~30000rpm,更优选为25000rpm,所述均质的时间优选为30~90s,更优选为60~90s。在本发明中,所述过筛的筛网尺寸优选为40目,取筛下部分进行后续的提取。
51.在本发明中,所述待测饲料的质量与乙腈的体积之比优选为1g:5~10ml,更优选为1g:5~8ml。
52.在本发明中,所述混合优选为涡旋混合,所述涡旋混合的速度优选≥2500rpm,更优选为2500~3000rpm;所述涡旋混合的时间优选≥30s,更优选为30~60s,更优选为30~40s。
53.在本发明中,所述超声提取的功率优选≥600w,更优选为600~1000w;所述超声提取的温度优选为40~45℃,更优选为43~45℃;所述超声提取的时间优选≥30min,更优选为30~40min,进一步优选为30~35min。
54.完成所述提取后,本发明优选还包括将所得提取体系进行离心分离,将所得上清液干燥,得到提取物。在本发明中,所述离心分离的速度优选≥6000rpm,更优选为6000~8000rpm;所述离心分离的时间优选为10~15min,更优选为10~13min。在本发明中,所述干燥优选为氮吹,所述氮吹的温度优选为40~45℃,更优选为40~42℃,本发明对于所述氮吹的时间没有特殊限定,氮吹至恒重即可。
55.在本发明中,所述待测饲料的质量与甲醇的体积之比优选为1g:0.5~1ml,更优选为1g:0.8~1ml。
56.得到甲醇复溶液后,本发明将所述甲醇复溶液依次进行吸附净化和正己烷洗涤,得到真菌毒素提取液。
57.在本发明中,所述真菌毒素包括交链孢酚、交链孢烯、玉米赤霉烯酮、交链孢毒素ii和腾毒素中的一种或几种。
58.在本发明中,所述吸附净化用优选包括c18吸附剂和psa吸附剂;所述c18吸附剂的粒径优选为40~50μm;所述psa吸附剂优选为乙二胺-n-丙基硅烷化硅胶,所述psa吸附剂的粒径优选为40~60μm;当所述吸附剂为c18吸附剂和psa吸附剂的混合物时,所述吸附剂中c18吸附剂和psa吸附剂的质量比优选为1:0.4~0.6,更优选为1:0.5~0.55。在本发明中,所述待测饲料和吸附剂的质量比优选为1:0.2~0.6,更优选为1:0.3~0.5。
59.在本发明中,所述吸附净化优选为涡旋混合,所述涡旋混合的速度优选≥2500rpm,更优选为2500~3000rpm;所述涡旋混合的时间优选≥30s,更优选为30~60s,进一步优选为30~40s。
60.完成所述净化后,本发明优选还包括将所得净化体系进行离心分离,将所得上清液(即甲醇净化液)进行正己烷洗涤。在本发明中,所述离心分离的速度优选≥6000rpm,更优选为6000~8000rpm;所述离心分离的时间优选为10~15min,更优选为10~13min。在本发明中,所述待测饲料的质量与正己烷的体积之比优选为1g:0.5~2ml,更优选为1g:0.5~1ml。
61.在本发明中,所述正己烷洗涤优选包括:在所得甲醇净化液中加入正己烷依次进行涡旋混合和分层。在本发明中,所述涡旋混合的速度优选≥2500rpm,更优选为2500~3000rpm;所述涡旋混合的时间优选≥30s,更优选为30~60s,进一步优选为30~40s;所述分层优选为离心分离,所述离心分离的速度优选≥6000rpm,更优选为6000~8000rpm;所述离心分离的时间优选为10~15min,更优选为10~13min。
62.本发明还提供了一种饲料中真菌毒素的检测方法,包括以下步骤:对上述技术方案所述提取方法得到的真菌毒素提取液进行液相色谱串联质谱检测。
63.在本发明中,所述真菌毒素包括交链孢酚、交链孢烯、玉米赤霉烯酮、交链孢毒素ii和腾毒素中的一种或几种。本发明优选先将所述真菌毒素提取液过0.22μm滤膜后再进行液相色谱串联质谱检测。
64.在本发明中,所述液相色谱的检测条件包括:色谱柱优选为c18柱,更优选为beh c
18
柱(100mm
×
2.1mm,1.7μm);柱箱温度优选为40℃;进样量优选为5μl;流动相体系优选为
流动相a和流动相b,所述流动相a优选为甲醇,所述流动相b优选为水;所述流动相体系的流速优选为0.3ml/min;洗脱方式优选为梯度洗脱;
65.所述梯度洗脱的程序优选如表1所示:
66.表1梯度洗脱程序
[0067][0068][0069]
即,0.00~2.00min:所述流动相a的体积百分含量为90%;
[0070]
2.00~4.00min:所述流动相a的体积百分含量由10%匀速增加到95%;
[0071]
4.00~8.00min:所述流动相a的体积百分含量为95%;
[0072]
8.00~8.10min:所述流动相a的体积百分含量由95%匀速减少到10%;
[0073]
8.10~10.00min:所述流动相a的体积百分含量为10%。
[0074]
在本发明中,所述质谱的检测条件包括:离子源优选为电喷雾离子源;检测方式优选为多反应监测;扫描方式优选为负离子模式;喷雾电压优选为3.0kv;离子传输管温度优选为320℃;脱溶剂气温度优选为300℃;毛细管温度优选300℃;鞘气优选为氮气,所述鞘气的流速优选为40arb;辅助气优选为氩气,所述辅助气的流速优选为10arb;碰撞气优选为氩气(ar)。
[0075]
在本发明中,真菌毒素的定性离子对、定量离子对、碰撞能量、保留时间如表2所示。
[0076]
表2真菌的质谱参数
[0077][0078]
在本发明中,所述质谱检测采用的仪器优选为三重四级杆质谱(tsq endura)。
[0079]
在本发明中,所述高效液相色谱串联质谱对所述真菌毒素提取液进行检测优选包括定性检测和定量检测。
[0080]
本发明采用多反应监测(mrm)的方式对真菌毒素提取液进行定性和定量分析,其原理是在三重四级杆串联质谱中,通过第一个四级杆(q1)对母离子进行选择,在第二个四级杆(q2)中进行碰撞解离,通过第三个四级杆(q3)对子离子进行选择离子检测,只对符合特定条件的离子进行检测,该模式能够有效排除本底干扰,具有较高的选择性、信噪比和灵敏度。
[0081]
采用蠕动泵直接进样的方式进行质谱检测条件的优化,分别将5种霉菌毒素标准品配制成1.00μg/ml的溶液,在esi-模式下进行一级质谱扫描,选择合适的母离子;然后再分别进行二级质谱分析,找出在二级质谱中两个信号较强的碎片离子,将信号最强的碎片离子作为定量离子,另一个碎片离子作为辅助定性离子;同时对锥孔电压、去溶剂气流速、喷雾电压等参数进行优化,得到完整的质谱条件。
[0082]
在本发明中,所述定性检测的步骤优选包括:
[0083]
将所述真菌毒素提取液和混合标准工作液按照液相色谱检测的条件和质谱检测的条件进行测定,记录真菌毒素提取液和混合标准工作液中真菌的色谱保留时间,当真菌毒素提取液中检出与某标准工作溶液中的真菌标准品保留时间一致的色谱峰(变化范围在
±
2.5%之内),并且真菌毒素提取液mrm谱图中所选择的监测离子对的相对丰度比与相当浓度标准溶液的离子相对丰度比(k)的偏差不超过表3规定的范围,可以确定试样中检出相应化合物。
[0084]
表3为定性时相对离子丰度的最大允许偏差。
[0085]
表3定性时相对离子丰度的最大允许偏差
[0086]
相对离子丰度/%>5020-5010-20≤10
允许相对偏差/%
±
20
±
25
±
30
±
50
[0087]
在本发明中,所述混合标准工作液的配制方法优选包括:
[0088]
配制浓度为1.0mg/ml的标准储备溶液;
[0089]
配制浓度为10.0μg/ml的标准混合中间溶液;
[0090]
配制混合标准工作液。
[0091]
在本发明中,所述标准储备溶液的配制方法优选包括以下步骤:分别精密称真菌毒素标准品各10.0mg(精确至0.01mg),用甲醇溶解并稀释定容至10ml,摇匀,得到浓度为1.0mg/ml的标准储备溶液,-20℃避光密封保存。
[0092]
在本发明中,所述真菌毒素标准品优选为交链孢酚标准品、交链孢烯标准品、玉米赤霉烯酮标准品、交链孢毒素ii标准品和腾毒素标准品的具体规格优选详见表4,所述真菌毒素标准品的纯度独立地优选≥98%。
[0093]
表4真菌标准品的具体规格
[0094]
英文名中文名分子式cas号分子量交链孢酚(aoh)alternariolc
14h10
o5641-38-3258.23交链孢烯(alt)altenuenec
15h16
o629752-43-0292.28玉米赤霉烯酮(zen)zearalenonec
18h22
o517924-92-4318.36交链孢毒素ii(atx ii)altertoxins iic
20h14
o656257-59-1350.32腾毒素(ten)tentoxinc
22h30
n4o428540-82-1414.5
[0095]
在本发明中,所述混合标准中间工作溶液的配制方法优选包括以下步骤:分别准确吸取标准储备溶液,用甲醇稀释定容,摇匀,制成真菌毒素浓度均为10.0μg/ml的标准混合中间溶液,4℃避光密封保存。本发明中对吸取标准储备溶液的体积以及利用甲醇稀释定容的最终体积没有特殊要求,以最终能够得到相应浓度的标准混合中间溶液为准。
[0096]
在本发明中,混合标准工作液的配制方法优选包括以下步骤:分别准确吸取标准混合中间溶液,用乙腈稀释定容,摇匀,作为混合标准工作液,得到浓度为1.0μg/ml的混合标准工作液,于4℃避光密封保存,有效期为2周。本发明中对吸取标准混合中间溶液的体积以及利用乙腈稀释定容的最终体积没有特殊要求,以最终能够得到相应浓度的混合标准工作液为准。
[0097]
在本发明中,所述混合标准工作液优选按照仪器响应情况配制或根据实际需要采用空白基质提取液,配制适当浓度的混合标准工作液。本发明中,所述空白基质提取液是指不含有上述真菌毒素的待测饲料,预处理步骤不变。
[0098]
本发明中,所述定量检测优选包括以下步骤:
[0099]
绘制标准曲线;
[0100]
根据所述标准曲线得到真菌毒素提取液中真菌毒素含量。
[0101]
在本发明中,所述标准曲线的绘制方法,优选包括以下步骤:
[0102]
将空白饲料按照前述待测饲料的前处理方式(提取和净化)进行处理,得到空白基质溶液;
[0103]
利用空白基质溶液对所述混合标准工作液进行逐级稀释,得到系列基质标准工作溶液;
[0104]
将所述系列基质标准工作溶液中的真菌毒素进行液相色谱串联质谱检测,得到真
菌毒素的色谱峰面积,以系列基质标准工作溶液的浓度为横坐标,真菌毒素的定量离子的色谱峰的峰面积为纵坐标,绘制标准曲线,得到标准曲线回归方程。
[0105]
在本发明中,所述系列基质标准工作溶液中真菌毒素的浓度分别为1.0μg/l、5.0μg/l、10.0μg/l、50.0μg/l和100μg/l。
[0106]
在本发明中,所述液相色谱串联质谱检测的条件优选与前述待测饲料的液相色谱串联质谱检测条件相同,在此不再赘述。
[0107]
得到标准曲线后,本发明根据所述标准曲线得到真菌毒素提取液中真菌毒素含量。
[0108]
在本发明中,所述待测饲料中真菌毒素含量的计算式,如式(1)所示:
[0109][0110]
x—试料中真菌毒素的残留量,单位为微克每千克(μg/kg);
[0111]
a—试料溶液中被测组分的色谱峰面积;
[0112]as
—标准工作溶液中被测组分的色谱峰面积;
[0113]cs
—标准工作溶液中被测组分的浓度,单位为微克每升(μg/l);
[0114]
v—试料最终定容体积,单位为毫升(ml);
[0115]
m—供试试料质量,单位为克(g);
[0116]
计算结果需扣除空白值,测定结果用平行测定的算术平均值表示,保留三位有效数字。
[0117]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0118]
实施例1
[0119]
标准曲线的绘制
[0120]
标准储备溶液(1.0mg/ml):分别准确称取适量5种真菌毒素标准品(aoh标准品、alt标准品、zen标准品、atx ii标准品和ten标准品)置于10ml容量瓶中,用甲醇溶解并稀释至刻度,混匀,配制成浓度为1mg/ml的标准储备溶液,于-20℃条件下避光密封保存。
[0121]
标准混合中间溶液(10.0μg/ml):分别准确移取适量5种真菌毒素标准储备溶液,置于100ml容量瓶中,用乙腈稀释至刻度,混匀,配制成5种真菌毒素浓度均为10.0μg/ml的混合标准中间溶液,于4℃条件下避光密封保存。
[0122]
混合标准工作液(1.0μg/ml):准确移取适量标准混合中间溶液,置于10ml容量瓶中,用乙腈溶解并稀释至刻度,混匀,配制成5种真菌毒素浓度均为1.0μg/ml的混合标准工作液,于4℃避光密封保存,有效期为2周。
[0123]
空白基质溶液的制备的配制:称取2.0g(精确至
±
0.01g)空白渔用饲料(不含真菌毒素)于50ml离心管中,加入乙腈10ml,在3000rpm条件下涡旋混合30s,在45℃、600w条件下超声提取30min,在6000rpm条件下离心分离10min,准确移取上清液5ml于15ml离心管,在40℃条件下氮吹至无乙腈残留,用甲醇复溶并定容至1ml,得到甲醇复溶液。在所述甲醇复溶液中加入0.4g粒径为40~50μm的c18吸附剂和0.2g粒径为40~60μm的乙二胺-n-丙基硅烷
化硅胶,在3000rpm条件下涡旋混匀30s,在6000rpm条件下离心分离10min,转移上清液,在上清液中加入正己烷1ml,在3000rpm条件下涡旋混匀30s,在6000rpm条件下离心分离10min,弃上层正己烷层,下层甲醇层过0.22μm滤膜,得到空白基质溶液;
[0124]
系列基质标准工作溶液的配制:利用空白基质溶液对所述混合标准工作液进行逐级稀释,得到真菌毒素的浓度分别为1.0μg/l、5.0μg/l、10.0μg/l、50.0μg/l和100μg/l的系列基质标准工作溶液;
[0125]
将所述系列基质标准工作溶液中的真菌毒素进行液相色谱串联质谱检测,得到真菌毒素的色谱峰面积,以系列基质标准工作溶液的浓度为横坐标,真菌毒素的定量离子的色谱峰的峰面积为纵坐标,绘制标准曲线,得到如表5所示的标准曲线回归方程。
[0126]
其中,液相色谱的检测条件包括:色谱柱为beh c
18
柱(100mm
×
2.1mm,1.7μm);柱箱温度为40℃;进样量为5μl;流动相体系为流动相a和流动相b,所述流动相a为甲醇,所述流动相b为水;所述流动相体系的流速为0.3ml/min;洗脱方式为梯度洗脱,梯度洗脱的程序优选如表1所示。
[0127]
质谱的检测条件包括:采用三重四级杆质谱仪(tsq endura),离子源为电喷雾离子源;检测方式为多反应监测;扫描方式为负离子模式;喷雾电压为3.0kv;离子传输管温度为320℃;脱溶剂气温度为300℃;毛细管温度300℃;鞘气(氮气,流速为40arb);辅助气(氩气,流速为10arb);碰撞气为氩气(ar);真菌毒素的定性离子对、定量离子对、碰撞能量、保留时间如表2所示。
[0128]
表5真菌毒素的标准曲线回归方程
[0129] 标准曲线回归方程r2alty=-5299.07+2092.61*x0.9967alx
ꢀⅱ
y=28816.8+7009.92*x0.9983aohy=24213.2+6340.27*x0.9953teny=3012.67+2820.89*x0.9971zeny=30550+11569.7*x0.9979
[0130]
说明,本发明提供的检测方法,5种真菌毒素在各自的响应范围内线性关系良好,相关系数(r2)均大于0.99。
[0131]
实施例2
[0132]
提取溶剂的选择
[0133]
(1)加标样品的配制:称取2.0g(精确至
±
0.01g)空白渔用饲料(不含真菌毒素),经25000rpm条件下均质70s过40目筛,将筛下部分的空白渔用饲料于50ml离心管中,加入20μl实施例1配制的混合标准工作液,在3000rpm条件下涡旋混匀30s,静置30min,得到加标样品。共平行制备6份加标样品。
[0134]
(2)提取:分别在6份加标样品中加入10ml不同的提取溶剂(乙腈、甲醇、1%(v/v)甲酸-乙腈混合溶剂、1%甲酸(v/v)-甲醇混合溶剂、70%(v/v)乙腈水溶液和84%(v/v)乙腈水溶液),然后在3000rpm条件下涡旋混合30s,置于超声水浴锅中,在45℃、600w条件下超声提取30min,在6000rpm条件下离心分离10min,准确移取5ml上清液于15ml离心管,在40℃条件下氮吹至干,用甲醇复溶并定容至1ml,得到甲醇复溶液。
[0135]
(3)净化:在所述甲醇复溶液中加入0.4g粒径为40~50μm的c18吸附剂和0.2g粒径
为40~60μm的乙二胺-n-丙基硅烷化硅胶,在3000rpm条件下涡旋混匀30s,在6000rpm条件下离心分离10min,转移上清液,在上清液中加入正己烷1ml,在3000rpm条件下涡旋混匀30s,在6000rpm条件下离心分离10min,弃上层正己烷层,下层甲醇层过0.22μm滤膜,得到待测加标样品溶液。
[0136]
(4)检测:按照实施例1的液相色谱串联质谱检测条件对加标样品和待测加标样品溶液进行中的真菌毒素进行液相色谱串联质谱检测,真菌毒素的回收率如表6和图1~图2所示,其中,图1为加标样品溶液mrm谱图;
[0137]
图2为不同提取剂下真菌毒素的回收率结果图。
[0138]
表6不同提取剂下真菌毒素的回收率(%)
[0139] altaltoxaohtenzen乙腈92.285.578.693.590.2甲醇85.974.467.287.176.81%甲酸-乙腈91.580.281.490.387.21%甲酸-甲醇87.965.768.381.880.570%乙腈水溶液82.081.177.786.285.684%乙腈水溶液85.583.672.588.187.9
[0140]
由图1~图2和表6可知,当采用乙腈为提取溶剂时多种霉菌毒素的回收率均能达到比较理想的效果。
[0141]
实施例3
[0142]
提取技术的选择与最优化
[0143]
按照实施例2的方法测试真菌毒素的回收率,与实施例2的区别在于:制备3份加标样品,分别记为第1组、第2组和第3组,在3组加标样品中加入乙腈(第1组加入20ml,第2组和第3组均加入10ml),采用不同提取方式(组1:采用图3所示的振荡器,在室温、320rpm条件下振荡提取2h;组2:采用图4所示的均质器,在室温、30000rpm条件下均质提取30s;组3:在3000rpm条件下涡旋混合30s,然后在45℃、600w条件下超声提取30min)进行提取,在6000rpm条件下离心分离10min,准确移取上清液(第1组移取10ml,第2组和第3组均移取5ml)于15ml离心管,在40℃条件下氮吹至干,用甲醇复溶并定容至1ml,得到甲醇复溶液。真菌毒素的回收率如图5和表7所示。
[0144]
表7不同提取方式下真菌毒素回收率(%)
[0145] altaltoxaohtenzen超声提取91.586.980.591.693.5振荡提取87.782.181.286.296.3均质提取88.690.586.991.590.7
[0146]
由图5和表7可知,3种提取方式均可获得比较理想的提取效果,其中采用振荡提取所需时间较长,且所需提取试剂多;采用均质提取时单个样品所需时间最短,但一次只能处理一个样品,无法实现高通量,因此在进行批量检测时,总体提取时间会显著增加,不利于提高提取时间效率。而采用超声提取时,多种真菌毒素的回收率、提取效果及样品间提取效果的平行性可达到最理想效果,且超声水浴锅至少可实现24个样品的同时提取,处理效率高。
[0147]
实施例4
[0148]
净化技术的选择与最优化
[0149]
将属于quechers技术的c18吸附剂、psa吸附剂与基于液液萃取原理的正己烷净化方法联合使用。对联合使用效果进行验证,共设3个组:第1组为c18和正己烷联合;第2组是psa(乙二胺-n-丙基硅烷化硅胶)和正己烷联合;第3组是c18、psa(乙二胺-n-丙基硅烷化硅胶)和正己烷的联合,具体如下:
[0150]
按照实施例2的方法测试真菌毒素的回收率,与实施例2的区别在于:制备3份加标样品,分别记为第1组、第2组和第3组;净化过程中,第1组:在1ml甲醇复溶液中加入0.6g c18吸附剂;第2组:在1ml甲醇复溶液中加入0.6g psa吸附剂;第3组:在1ml甲醇复溶液中加入0.4g c18吸附剂和0.2g psa吸附剂。不同净化条件下真菌毒素的回收率如图6~图8和表8所示,其中,图6为c18吸附剂和正己烷联合净化的mrm谱图,图7为psa吸附剂和正己烷联合净化的mrm谱图,图8为c18吸附剂、psa吸附剂和正己烷联合净化的mrm谱图。
[0151]
表8不同净化条件下真菌毒素的回收率(%)
[0152] altaltoxaohtenzenc18+正己烷83.684.276.987.489.4psa+正己烷92.588.983.490.292.9c18+psa+正己烷93.187.981.289.692.4
[0153]
由表8和图6~图8可知,综合各实验条件下的净化效果,将0.4g c18吸附剂(粒径40μm~50μm)、0.2g psa吸附剂(乙二胺-n-丙基硅烷化硅胶,粒径40~60μm)与正己烷净化联合使用,先经c18和psa混合吸附剂净化,将c18吸附剂和psa吸附剂优势范围内的大部分色素、脂类及其它杂质很好的去除后,后加入正己烷进一步去除脂类等弱极性杂质,多种毒素的回收率和净化效果均达到满意效果。
[0154]
实施例5
[0155]
液相色谱检测条件的优化
[0156]
色谱柱优化:将实施例1中配制的浓度为10.0μg/l的5种真菌毒素的基质标准混合溶液进行液相色谱串联质谱检测,其中,色谱柱分别为beh c
18
(100mm
×
2.1mm,1.7μm)、hss t3(100mm
×
2.1mm,1.8μm)和hss c
18
(100mm
×
2.1mm,1.7μm),其他检测条件与实施例1相同,发现beh c
18
(100mm
×
2.1mm,1.7μm)色谱柱对5种霉菌毒素的分离效果及峰形最好,因此,选择beh c
18
(100mm
×
2.1mm,1.7μm)色谱柱作为色谱分离柱。
[0157]
流动相体系优化:将5种标准溶液进行液相色谱串联质谱检测,其中,流动相a(有机相)和流动相b(水相)组成的流动相体系分别为乙腈和0.1%(v/v)甲酸水溶液、乙腈和0.1%(v/v)甲酸-乙酸铵水溶液(乙酸铵浓度为5mmol/l)、乙腈和水、甲醇和0.1%(v/v)甲酸-乙酸铵水溶液(乙酸铵浓度为5mmol/l)、甲醇和0.1%(v/v)甲酸水溶液、甲醇和水,其他检测条件与实施例1相同,5种标准溶液的mrm谱图如图9~图14所示,其中,图9为乙腈和0.1%(v/v)甲酸水溶液作为流动相时的mrm谱图,图10为乙腈和0.1%(v/v)甲酸-乙酸铵水溶液(乙酸铵浓度为5mmol/l)作为流动相时的mrm谱图,图11为乙腈和水作为流动相时的mrm谱图,图12为甲醇和0.1%(v/v)甲酸-乙酸铵水溶液(乙酸铵浓度为5mmol/l)作为流动相时的mrm谱图,图13为甲醇和0.1%(v/v)甲酸水溶液作为流动相时的mrm谱图,图14为甲醇和水作为流动相时的mrm谱图。由图9~图14可知,以beh c
18
(100mm
×
2.1mm,1.7μm)为色
谱柱,以甲醇和水作为流动相,五种真菌毒素分离度好、响应值高,满足残留检测要求。
[0158]
实施例6
[0159]
(1)灵敏度检测
[0160]
采用在空白基质中加标的方法来确定5种真菌毒素的定量限,具体步骤如下:在空白渔用饲料(不含真菌毒素)中添加实施例1配制的混合标准工作液,按照实施例1的前处理条件法进行提取和净化,以信噪比(s/n≥3)确定检出限(lod),以信噪比(s/n≥10)确定定量限(loq),得到5种真菌毒素的检出限均为1.0μg/kg,定量限为均2.0μg/kg。
[0161]
(2)稳定性
[0162]
在2.0g空白渔用饲料中加入混合标准工作液,加标浓度为10.0μg/kg,设置6个平行试验,测定3个批次,测试结果如表9所示
[0163]
表9稳定性测试结果
[0164][0165][0166]
由表9可知,平均加标回收率75.1%~87.3%(准确度,n=6,满足60%~120%要求),相对标准偏差(变异系数,rsd)3.33%~9.05%(稳定性,n=6,满足≤15%要求)。说明,本发明提供方法的提取和净化效果好,灵敏度高、准确性和稳定性好,适合于渔用饲料样品中真菌毒素的多残留、高通量检测。
[0167]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应
视为本发明的保护范围。
- 该技术已申请专利。仅供学习研究,如用于商业用途,请联系技术所有人。
- 技术研发人员:刘慧慧 姜立生 张华威 张秀珍 黄会 王艺华 李焕霞 崔庆奎 薛敬林 韩典峰 田秀慧 丁玉竹 孙琰晴 崔艳梅
- 技术所有人:栖霞市检验检测中心 烟台富美特信息科技股份有限公司
- 我是此专利的发明人
- 该领域下的技术专家
- 如您需求助技术专家,请点此查看客服电话进行咨询。
- 1、张老师:1.探索新型氧化还原酶结构-功能关系,电催化反应机制 2.酶电催化导向的酶分子改造 3.纳米材料、生物功能多肽对酶-电极体系的影响4. 生物电化学传感和生物电合成体系的设计与应用。
- 2、邬老师:1.高分子材料的共混与复合 2.涉及材料功能化及结构与性能的研究; 高分子热稳定剂的研发
- 3、褚老师:高分子生物材料与生物传感器,包括抗菌/抗污高分子材料、生物基高分子材料、超分子水凝胶、蛋白质材料的合成与自组装、等离子体聚合功能薄膜、表面等离子体共振光谱(SPR)、表面增强拉曼散射(SERS)生物传感器等。
- 4、廖老师:1. 晶面可控氧化铝、碳基载体及催化剂等高性能、新结构催化材料研究 2. 乙烯环氧化催化剂的研究与开发 3. 低碳不饱和烯烃的选择性氧化催化剂及工业技术开发
- 5、李老师:1. 加氢精制 2. 选择加氢 3. 加氢脱氧 4. 介孔及介微孔分子筛合成及催化应用
- 如您是高校老师,可以点此联系我们加入专家库。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
精彩留言,会给你点赞!
专利分类正在加载中....