一种选择性加氢脱除间戊二烯中炔烃和环戊二烯的催化剂及其制备方法
1.本发明属于催化剂技术领域,涉及一种选择性加氢脱除间戊二烯中炔烃和环戊二烯的催化剂及其制备方法。
背景技术:
2.近年来,随着我国乙烯生产规模的不断扩大及以液态烃为原料制取乙烯生产能力的不断增长,碳五(c5)馏分产量日趋丰富。c5馏分的组成十分复杂,主要为含有4-6个碳原子的烷烃、烯烃、环烷烃、环烯烃、双烯烃和炔烃等组成的混合烃类。其中含量及利用价值较高的组分为异戊二烯(isp,质量分数为15%-20%)、间戊二烯(pd,质量分数为10%-19%)、环戊二烯(cpd,质量分数为14%-19%)。这些二烯烃是生产异戊橡胶、丁基橡胶、乙丙橡胶、石油树脂、热塑性弹性体和不饱和聚酯等石油化工和精细化工产品的重要原料。其中,间戊二烯是用于生产脂肪族石油树脂的主要原料,这种石油树脂的分子链节主要是脂肪烃结构,酸值低、粘结性能好、耐水耐乙醇、耐化学品腐蚀、相容性好,已逐步应用到压敏胶粘剂、浅色热熔胶、粉末涂料、浅色热熔性标识漆、油漆以及印刷油墨添加剂等领域。间戊二烯作为生产石油树脂的原料,需要从c5馏分中抽提出来,而c5馏分中含有的少量炔烃(如戊炔-1、戊炔-2和异丙烯基乙炔等)和环戊烯等,其总含量约为0.1-2 wt.%,由于它们的化学性质活泼,对间戊二烯的聚合过程产生严重的影响(使引发剂失活,导致难以聚合)。因此,以间戊二烯为原料进行聚合生产石油树脂等聚合物前需要将其中的炔烃和环戊二烯等杂质脱除。
3.目前,脱除烯烃中少量炔烃和环戊二烯的方法主要有萃取精馏法和选择性加氢法。其中,萃取精馏法增加了被分离组分之间的相对挥发度,使c5馏分沸点相近的组分得以分离。但该方法存在工艺流程复杂,能耗高等缺点。并在分离过程中炔烃会在某些设备中浓缩,产生不安全因素,在吸收过程中有机溶剂易挥发,对环境造成污染,限制了其在工业中的应用。相比较而言,选择性加氢法能够有效脱除烯烃中的炔烃和环戊二烯,提高烯烃的收率,且工艺流程简单、能耗低和环境友好,是目前脱除烯烃中炔烃和环戊二烯应用最广泛的技术。选择性加氢法脱除炔烃和环戊二烯的核心是选择性加氢催化剂的制备,与ni、co和fe等普通金属相比,贵金属pd具有优异的低温催化活性和良好的选择性加氢活性,常用的选择性加氢脱除烯烃中炔烃和环戊二烯的催化剂以pd为活性金属,将其负载于γ-a12o3或sio2等惰性载体上。但由于贵金属pd在载体上的分散性较差,导致其负载量较高,使得其制备成本较高。针对此问题,卫国宾等(卫国宾,杨思源,张敬畅,等.石油化工,2012,41(11):1239-1244.)采用微乳法制备了pd纳米粒子,并将其负载于γ-a12o3上,制备了高分散的pd催化剂,该催化剂在乙炔选择性加氢体系中,能够有效脱除其中的乙炔,但该催化剂对乙烯的选择性较差。为了克服现有技术中加氢催化剂脱炔工艺中存在的烯烃选择性差的缺陷。cn111097445a公开了一种异戊二烯选择性加氢除炔的方法,该催化剂以钯为活性组分,铜为助剂,氧化铝为载体。该方法制备的催化剂在异戊二烯选择性加氢除炔反应中表现出优异的催化活性和选择性,总剩炔量为10 μg/g,异戊二烯的损失率为1.0%。虽然其异戊二烯
损失率较低,但是炔烃没有完全脱除,仍然会导致后续聚合过程中引发剂的失活,影响聚合产品的质量;并且,其反应采用两段反应器,增加了设备投入。
4.由上述可知,目前报道较多的是针对c
2-c4单烯烃馏分中少量二烯烃或炔烃选择性加氢催化剂以及少量c5馏分中异戊二烯的选择性加氢脱炔催化剂的开发,但是,对于间戊二烯中少量炔烃和环戊二烯的脱除,其相关选择性加氢催化剂的制备尚未见报道。
5.综上所述,本领域亟需开发一种针对脱除间戊二烯中少量炔烃和环戊二烯的选择性加氢催化剂及其制备方法。
技术实现要素:
6.为解决上述技术问题,本发明的目的是提供了一种选择性加氢脱除间戊二烯中炔烃和环戊二烯的催化剂及其制备方法,该催化剂具有优异的加氢选择性,能够有效脱除间戊二烯中的炔烃和环戊二烯,并使间戊二烯具有较低的损失率。
7.为了达到上述目的,本发明提供了一种选择性加氢脱除间戊二烯中炔烃和环戊二烯的催化剂及其制备方法,该方法包括以下步骤:(1)配置含有钯化合物的水溶液,将氯化钾加入其中得到溶液a,再向a溶液中加入一元醇和非离子表面活性剂,在一定温度下搅拌一段时间,得到溶液b;(2)以sio2为载体并将其悬浮于水中,加入一定量的两性表面活性剂得到悬浮液c;(3)将溶液b逐滴加入悬浮液c中,在一定温度下搅拌一段时间,得到悬浮液d;(4)配置含有铜化合物的水溶液,加入一定量的沉淀剂得到浊液e;(5)将浊液e逐滴加至悬浮液d中,在一定温度下搅拌一段时间后,过滤、洗涤、干燥和焙烧,得到氧化态催化剂;(6)将氧化态催化剂还原,得到选择性加氢脱除间戊二烯中炔烃和环戊二烯的催化剂。在上述制备方法的步骤(1)中,优选地,所述钯化合物为醋酸钯、氯化钯或硝酸钯;更优选地,所述钯化合物水溶液的浓度为0.001 mol/l-0.010 mol/l;钯原子与氯原子的摩尔比为1:(1-5)。
8.在上述制备方法的步骤(1)中,优选地,所述一元醇为甲醇、乙醇、正丙醇或异丙醇;优选地,一元醇与去离子水的体积比为1:(0.5-5)。
9.在上述制备方法的步骤(1)中,优选地,所述非离子表面活性剂为聚乙烯亚胺、聚丙烯酸或聚乙烯吡咯烷酮;更优选地,非离子表面活性剂的浓度为0.010 mol/l-0.200 mol/l,钯原子与非离子表面活性剂的摩尔比为1:(10-30)。
10.在上述制备方法的步骤(1)中,优选地,所述反应的反应温度为40-70 ℃,反应时间为4-8 h。
11.在上述制备方法的步骤(2)中,优选地,所述两性表面活性剂为脂肪酸甲酯乙氧基化物的磺酸盐、十二烷基二甲基甜菜碱、椰油酰胺丙基羟磺酸甜菜碱、月桂酰胺基丙基甜菜碱和谷胱甘肽中的一种或几种;更优选地,两性表面活性剂的浓度为0.001 mol/l-0.050 mol/l,铜原子与两性表面活性剂的摩尔比为1:(1-20)。
12.在上述制备方法的步骤(3)中,优选地,所述反应的反应温度为50-80 ℃,反应时间为8-16 h。
13.在上述制备方法的步骤(4)中,优选地,所述铜化合物为醋酸铜、氯化铜或硝酸铜;更优选地,所述铜化合物水溶液的浓度为0.0001 mol/l-0.0200 mol/l,钯原子与铜原子的摩尔比为1:(0.1-20)。
14.在上述制备方法的步骤(4)中,优选地,所述沉淀剂为碳酸钠、氢氧化钠、或尿素中的一种或几种;更优选地,所述沉淀剂水溶液的浓度为0.001 mol/l-0.100 mol/l,铜原子与沉淀剂的摩尔比为1:(10-30)。
15.在上述制备方法的步骤(5)中,优选地,所述干燥温度为100 ℃-150 ℃,干燥时间为12-24 h;所述焙烧的温度400 ℃-600 ℃,时间4 h-12 h;更优选地,焙烧气氛为氮气和氧气混合气,氮气:氧气体积比为5-1:1。
16.本发明提供了一种由上述制备方法制备得到的选择性加氢脱除间戊二烯中炔烃和环戊二烯催化剂,其用于间戊二烯选择性加氢脱除炔烃和环戊二烯反应的条件为温度25-70 ℃,液体体积空速为10-50 h-1
,压力为0.1-1 mpa,氢油比为10-50。
17.本发明的有益效果:(1)本发明所提供的选择性加氢脱除炔烃和环戊二烯催化剂经过两性表面活性剂的分散作用,提高了pvp包覆的pd纳米颗粒的分散性,避免其在焙烧过程中的团聚,从而增加了活性中心的数量,有利于氢在金属表面的吸附和扩散,提高了催化剂对炔烃和环戊二烯的反应的活性。
18.(2)本发明在两性表面活性剂分散的pd纳米颗粒中,通过不同沉淀剂定向引入cu对其修饰,降低了pd纳米颗粒中角位原子的数量,提高了边位pd原子的数量,从而提高了催化剂对间戊二烯的加氢选择性,降低了间戊二烯的饱和率,提高了间戊二烯的收率。
具体实施方式
[0019] 为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现对本发明的技术方案进行以下详细说明,但不能理解为对本发明的可实施范围的限定。实施例中,各原始试剂材料均可商购获得,未注明具体条件的实验方法为所属领域熟知的常规方法和常规条件,或按照仪器制造商所建议的条件。
[0020]
实施例1本实施例提供一种选择性加氢脱除间戊二烯中炔烃和环戊二烯催化剂的制备方法,该方法包括以下步骤:步骤一,分别量取10 ml浓度为0.004 mol/l氯化钯(pdcl2)和10 ml浓度为0.008 mol/l氯化钾(kcl),混合以后,加入0.09 g非离子表面活性剂聚乙烯吡咯烷酮(pvp),40 ℃搅拌4 h,得到含有pd化合物的溶液。
[0021]
步骤二,称取0.0158 g两性表面活性剂脂肪酸甲酯乙氧基化物的磺酸盐(fmes)溶解在20 ml去离子水中,搅拌均匀后,加入2 g载体sio2,搅拌4 h,得到sio2悬浮液,将其加入含有pd化合物的水溶液中,40 ℃搅拌12 h,得到pd-sio2悬浮液。
[0022]
步骤三,称取沉淀剂无水碳酸钠(na2co3)0.347 g溶于10 ml去离子水中,搅拌,待
其完全溶解后,量取10 ml浓度为0.008 mol/l硝酸铜(cu(no3)2∙
3h2o)加到该溶液中,40 ℃搅拌,得到含有cu化合物的溶液。
[0023]
步骤四,将含有cu化合物的溶液逐滴加入上述pd-sio2悬浮液中,继续搅拌3 h,得到cupd-sio2悬浮液。反应完成后过滤、洗涤,再将样品置于烘箱中120 ℃干燥12 h,最后在空气气氛中焙烧,得到氧化态选择性加氢催化剂。
[0024]
步骤五,将所得的氧化态催化剂还原,还原压力为1.5 mpa,氢气流量为20 ml/min,温度300 ℃,还原时间4 h,得到活化后的加氢催化剂。活化后的催化剂,其反应性能评价条件为温度30 ℃,液体体积空速为15 h-1
,压力为0.1 mpa,氢油比为10。
[0025]
实施例2步骤一,分别量取10 ml浓度为0.004 mol/l氯化钯(pdcl2)和10 ml浓度为0.008 mol/l氯化钾(kcl),混合以后,加入0.09 g非离子表面活性剂聚乙烯吡咯烷酮(pvp),40 ℃加热搅拌4 h,得到含有pd化合物的溶液。
[0026]
步骤二,称取0.008 g两性表面活性剂谷胱甘肽溶解在20 ml去离子水中,搅拌均匀后,加入2 g载体sio2,搅拌4 h,得到sio2悬浮液,将其加入含有pd化合物的水溶液中,40 ℃搅拌12 h,得到pd-sio2悬浮液。
[0027]
步骤三,称取沉淀剂无水碳酸钠(na2co3)0.347 g溶于10 ml去离子水中,搅拌,待其完全溶解后,量取10 ml浓度为0.008 mol/l硝酸铜(cu(no3)2∙
3h2o)加到该溶液中,40 ℃搅拌,得到含有cu化合物的溶液。
[0028]
步骤四,将含有cu化合物的溶液逐滴加入上述pd-sio2悬浮液中,继续搅拌3 h,得到cupd-sio2悬浮液。反应完成后过滤、洗涤,再将样品置于烘箱中120 ℃干燥12 h,最后在空气气氛中焙烧,得到氧化态选择性加氢催化剂。
[0029]
步骤五,将所得的氧化态催化剂还原,还原压力为1.5 mpa,氢气流量为20 ml/min,温度300 ℃,还原时间4 h,得到活化后的加氢催化剂。活化后的催化剂,其反应性能评价条件为温度30 ℃,液体体积空速为15 h-1
,压力为0.1 mpa,氢油比为10。
[0030]
实施例3步骤一,分别量取10 ml浓度为0.004 mol/l氯化钯(pdcl2)和10 ml浓度为0.008 mol/l氯化钾(kcl),混合以后,加入0.09 g非离子表面活性剂聚乙烯吡咯烷酮(pvp),40 ℃搅拌4 h,得到含有pd化合物的溶液。
[0031]
步骤二,称取0.0079 g两性表面活性剂十二烷基二甲基甜菜碱(bs-12)溶解在20 ml去离子水中,搅拌均匀后,加入2 g载体sio2,搅拌4 h,得到sio2悬浮液,将其加入含有pd化合物的水溶液中,40 ℃搅拌12 h,得到pd-sio2悬浮液。
[0032]
步骤三,称取沉淀剂无水碳酸钠(na2co3)0.347 g溶于10 ml去离子水中,搅拌,待其完全溶解后,量取10 ml浓度为0.008 mol/l硝酸铜(cu(no3)2∙
3h2o)加到该溶液中,40 ℃搅拌,得到含有cu化合物的溶液。
[0033]
步骤四,将含有cu化合物的溶液逐滴加入上述pd-sio2悬浮液中,继续搅拌3 h,得到cupd-sio2悬浮液。反应完成后过滤、洗涤,再将样品置于烘箱中120 ℃干燥12 h,最后在空气气氛中焙烧,得到氧化态选择性加氢催化剂。
[0034]
步骤五,将所得的氧化态催化剂还原,还原压力为1.5 mpa,氢气流量为20 ml/min,温度300 ℃,还原时间4 h,得到活化后的加氢催化剂。活化后的催化剂,其反应性能评
价条件为温度30 ℃,液体体积空速为15 h-1
,压力为0.1 mpa,氢油比为10。
[0035]
对比例本对比例提供一种选择性加氢脱除间戊二烯中炔烃和环戊二烯催化剂的制备方法,该方法包括以下步骤:称取0.0074 g氯化钯(pdcl2)溶解于盐酸(hcl)溶液中,等体积浸渍于2 g二氧化硅(sio2)粉末上,将所得样品于室温条件下搅拌24 h,120 ℃干燥12 h;再称取0.0399 g硝酸铜(cu(no3)2∙
3h2o)溶解于去离子水中,等体积浸渍于浸渍上pd的sio2上,将所得样品于室温条件下搅拌24 h,120 ℃干燥12 h,最后,在空气气氛中450 ℃焙烧4 h,得到参比催化剂。将所得的参比催化剂还原,还原压力为1.5 mpa,氢气流量为20 ml/min,温度300 ℃,还原时间4 h,以含有2-戊炔和环戊二烯的间戊二烯为原料,其反应性能评价条件为温度30 ℃,液体体积空速为15 h-1
,压力为0.1 mpa,氢油比为10。
[0036]
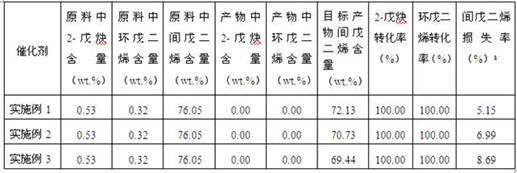
表1 实施例制备的催化剂的选择性加氢反应性能评价结果a(原料中间戊二烯含量-产物中间戊二烯含量)/原料中间戊二烯含量由表1可以看出,三种添加不同表面活性剂制备的选择性加氢催化剂中杂质2-戊炔、环戊二烯的转化率为100%,表明炔烃和环戊二烯已被完全脱除;其中使用表面活性剂fmes制备的催化剂,间戊二烯的损失率最少。
[0037]
表2 对比例制备的催化剂的选择性加氢反应性能评价结果a(原料中间戊二烯含量-产物中间戊二烯含量)/原料中间戊二烯含量由表2可以看出,使用浸渍法制备的催化剂,间戊二烯损失率较大,且其中杂质2-戊炔、环戊二烯没有被完全脱除。
[0038]
最后说明的是:以上实施例仅用于说明本发明的实施过程和特点,而非限制本发明的技术方法,尽管参照上述实施例对本发明进行了详细说明,本领域的普通技术人员应当理解:依然可以对本发明进行修改或等同替换,而不脱离本发明的精神和范围的任何修改或局部替换,均应涵盖在本发明的保护范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1