一种多孔金属氧化物催化剂及其制备方法和在等离子体催化体系中的应用
1.本发明涉及等离子体技术和催化化学技术领域,尤其涉及一种多孔金属氧化物催化剂及其制备方法和在等离子体催化体系中的应用。
背景技术:
2.等离子体催化技术由非热等离子体与催化剂耦合而成,具有反应温度低、活化能力强、启动迅速、设备简易等优点,在环境与能源领域表现出了极具潜力的应用前景。目前绝大多数等离子体催化领域研究均致力于构建高效实用型的等离子体催化系统,这本身也是等离子体催化研究领域试图攻克的关键难题之一。
3.基于贵金属纳米催化剂(如pt,pd与ru等)构建的等离子体催化体系通常能在反应中展现出较高的性能,但其在实际应用中依然存在着诸多难题和挑战,主要的困难来自两方面:
4.(1)等离子体与贵金属纳米催化剂间的协同还不够高效。在等离子体催化反应过程中,等离子体作用下的表面催化反应的贡献要高于气相反应,则能够与等离子体发生相互作用的催化活性位点的本征活性和密度自然成为影响反应性能的主导因素。将贵金属纳米化,从而在载体(以金属氧化物为主)表面构筑大量高活性的贵金属/氧化物界面体系是创制高效贵金属纳米催化剂的常用手段,但对于在载体表面实现贵金属纳米粒子的可控分散与稳定(控制纳米粒子的表面迁移聚集),并确保催化活性位点同等离子体的高效相互作用,目前尚缺乏有效方法。
5.(2)反应过程很难避免产生有害的放电副产物。实际工况下复杂的气体组成会使放电产生多种有害副产物(如o3与no
x
等)。为了改善等离子体催化反应的性能,通常需要提高放电强度,但这也会导致放电有害副产物浓度的增加,造成严重的二次污染。
技术实现要素:
6.针对现有技术存在的高效等离子体催化系统构建过程中遇到的上述难题,本发明的第一个目的在于提供一种多孔金属氧化物催化剂的制备方法,所述的制备方法能够获得负载有贵金属纳米粒子的多孔金属氧化物催化剂,能够改变催化剂表/界面场等的状态包括场分布特点、场协同特征、微放电特性等。
7.本发明的第二个目的在于提供一种负载有贵金属纳米粒子的多孔金属氧化物催化剂,所述多孔金属氧化物催化剂具有为等离子体催化反应营造独特的物理-化学环境,从而起到调控反应途径、调变反应物在活性位上的吸脱附行为以及分子内部化学键的作用。
8.本发明的第三个目的在于提供一种多孔金属氧化物催化剂协同等离子体系的构建方法,利用特殊结构形貌催化剂构筑与等离子体反应器定制间的协同控制,实现对等离子体催化系统内多物理与化学场的有效调变,在控制系统内多场高效协同耦合的同时,显著增强系统的运行稳定性。制备方法不仅能使构建的等离子体催化系统展现出高活性,而
且在用于反应时具有极其优异的稳定性。
9.为实现上述第一个目的,本发明提供了如下技术方案:一种多孔金属氧化物催化剂的制备方法,具体包括以下步骤:
10.s1:多孔金属氧化物的制备:将金属氧化物前驱体、贵金属纳米粒子、络合剂溶于去离子水中,搅拌均匀,得到金属前驱体溶液;其中,所述络合剂选自柠檬酸、柠檬酸铵、二乙醇胺;
11.s2:加入模板剂,采用浸渍方法将步骤s1得到的金属前驱体溶液吸附于模板剂表面,得到样品;所述金属前驱体溶液和模板剂的质量比为1:1;
12.s3:将步骤s2制得的样品于800-1100℃下焙烧,氧化除去模板剂,得到金属氧化物催化剂;
13.s4:将步骤s3制得的金属氧化物催化剂在还原性气氛下反应一段时间,使贵金属纳米粒子原位脱溶,组装于步骤s3制得的金属氧化物催化剂表面;
14.其中,贵金属纳米粒子原位脱溶的具体方法包括:还原性气氛下热处理或外场强化还原性气氛处理;
15.所述还原性气氛下热处理的方法包括:在还原气氛下,以0.5-1℃/min的升温速率升温至400-500℃,恒温反应一定时间,使得金属前驱体中的金属离子还原、贵金属纳米粒子原位析出、组装于步骤s3制得的金属氧化物催化剂表面;
16.所述外场强化还原性气氛处理的方法包括:在还原气氛下,采用等离子放电,放电功率为15w,放电时间为40min,使得金属前驱体中的金属离子还原、贵金属纳米粒子原位析出、组装于步骤s3制得的金属氧化物催化剂表面。
17.更进一步地,在步骤s3中,在将步骤s2制得的样品焙烧前,向步骤s2制得的样品中加入尿素、聚氧丙烯聚氧乙烯共聚物、乙醇以及乙二醇,混合均匀,然后水热合成24h。
18.通过采用上述技术方案,高温焙烧去除模板剂得到多孔金属氧化物催化剂,贵金属纳米粒子在多孔金属氧化物催化剂表面及孔道内部原位脱溶,构成大量贵金属纳米粒子/金属氧化物界面体系,催化剂的表面缺陷位及贵金属纳米粒子/金属氧化物界面体系所提供的化学场通过分解o3、降低氧俘获电子的活化能等途径,选择性地增强等离子区中o、oh、o
2-等活性物种的生成,多孔结构使放电通道均匀分布于结构孔道内,孔道内部等离子体以贵金属纳米粒子为中心沿孔道表面延展,有效增强了等离子体与催化活性位点的作用。
19.进一步地,所述的金属氧化物前驱体为可溶性金属盐;
20.所述金属氧化物前驱体选自la(no3)3·
6h2o、sr(no3)2·
6h2o、co(no3)
·
6h2o、mn(no3)2·
6h2o、fe(no3)2·
6h2o中的至少一种。
21.进一步地,所述贵金属纳米粒子选自pt、au、pd、ru中的至少一种。
22.进一步地,所述的模板剂选自软模板剂或硬模板剂;
23.所述模板剂选自pmma、ps、球形炭、n-甲基吡咯烷、介孔分子筛、表面活性剂中的任意一种。
24.更进一步地,所述表面活性剂选自二甲氧基四甘醇,聚乙二醇,乙二醇,乙二胺四乙酸,l-赖氨酸,二乙醇胺中的任意一种。
25.进一步地,所述络合剂、金属前驱体溶液、贵金属纳米粒子的摩尔比为1:1:0.3-0.8。
26.进一步地,在步骤s4中,所述的还原性气氛中氢气含量》3%或气氛中co含量》5%。更进一步地,所述还原性气氛选自h2/ar混合气、co/he混合气中的任意一种,所述h2/ar混合气中,所述h2含量为10-20wt%;所述co/he混合气中,所述co含量为5wt%。
27.为实现上述第二个目的,本发明提供了如下技术方案:一种由所述的制备方法制备得到的负载有贵金属纳米粒子的多孔金属氧化物催化剂。
28.为实现上述第三个目的,本发明提供了如下技术方案:一种多孔金属氧化物催化剂协同等离子体系的构建方法,将所述多孔金属氧化物催化剂放置于等离子体放电反应器的等离子体区后端或等离子体区内,使所述多孔金属氧化物催化剂与等离子体放电反应器原位耦合。
29.进一步地,所述等离子体放电反应器选自介质阻挡放电反应器、电晕放电反应器、滑动弧放电反应器中的任意一种;
30.进一步地,所述等离子体放电反应器选自平板式、筒式、滑动弧放电反应器的任意一种。
31.进一步地,所述等离子体放电反应器的供电电源选自交流电源或脉冲电源。更进一步地,当所述供电电源为交流电源时,频率为50hz-30khz;
32.当所述供电电源为脉冲电源时,脉宽《1000ns;电源施加电压处于2-8kv。
33.综上所述,本发明具有以下有益效果:
34.1、本发明通过功能性构筑合成负载有高活性贵金属纳米粒子的多孔金属氧化物催化剂;对于等离子体催化系统的特性调变可以从催化剂的多孔微纳结构、纳米贵金属/金属氧化物界面、等离子体与催化剂间相互作用等多个层次进行。
35.2、本发明在构建等离子体催化系统时将纳米贵金属/金属氧化物催化剂置于等离子区可灵活调变放电模式:一方面,催化剂独特的分级多孔结构使放电通道随机均匀地分布于微纳结构孔道内,且孔道内部等离子体会以贵金属纳米粒子为中心沿催化剂表面有效延展,这增强了等离子体与催化活性位点的作用,利于在催化剂表面定制物理-化学环境;另一方面,催化剂通过对dbd放电模式的调变,将传统的丝状强放电模式转变为均匀、无强电流脉冲的表面放电,避免了n2的快速解离与no
x
的生成;此外,贵金属纳米粒子/金属氧化物界面体系以及催化剂的表面缺陷位所提供的化学场还可通过分解o3、降低氧俘获电子的活化能等途径,选择性地增强等离子区中o、oh、o
2-等活性物种的生成。
36.3、本发明构建的等离子体催化系统利用等离子体在纳米贵金属/金属氧化物表面定制物理-化学环境,可以调控反应物在活性位上的吸脱附行为,削弱反应物分子化学键(增强活化能力),这在增强表面催化反应的同时,也能避免毒化物种对活性位点的吸附占据,提升等离子体催化反应连续高效运行的稳定性。
37.4、本发明采用灵活的方法将等离子体与催化剂耦合构建系统,装置的造价低、装配方式灵活多变,既适用于小规模的精细化工使用,也便于系统放大,实现规模化应用。
附图说明
38.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以
根据这些附图获得其他的附图。
39.图1为本发明实施例1和对比例1-2提供的等离子体系的微放电特性图;
40.图2为本发明实施例1和对比例1-2对甲苯转化率影响的结果及实施例1和对比例1-2对co2、co选择性影响的结果;
41.图3为本发明实施例2的au@lsc的sem图;
42.图4为本发明实施例2和对比例3-4的炭粉转化率及产物中co2的选择性的结果;
43.图5为本发明实施例3的ag@cemno
x
的48h催化活性图;
44.图6为本发明实施例3的滑动弧放电的催化活性图;
45.图7为本发明实施例3的ag@cemno
x
的sem图。
具体实施方式
46.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图1-7,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
47.本技术中所使用的原料和设备均可通过市售获得。
48.本技术中所涉及的球形炭模板均为球形活性炭。
49.原料和/或中间体的制备例
50.多孔金属氧化物催化剂的制备例1:
51.ru@lscfm催化剂的制备方法,具体包括以下步骤:
52.s1:称取2.5994g的la(no3)3,0.4233g的sr(no3)2,0.3659g的co(no3)2,0.4837g的fe(no3)3,2.0743g的rucl3、1.0737g的mn(no3)2溶入100ml去离子水中搅拌均匀,然后加入6.3042g的柠檬酸,柠檬酸能够有效络合金属盐离子,得到金属前驱体溶液;
53.s2:称取10ml步骤s1制得的金属前驱体溶液通过3-5次浸渍将溶液完全浸渍于2g的pmma模板的表面,然后以10000r/min进行离心处理,离心处理48h后分离得到样品;
54.s3:将步骤s2得到的样品置于马弗炉内焙烧8h(850℃,升温速率0.5℃/min),使样品发生自蔓延燃烧,氧化除去pmma模板剂,得到la
0.8
sr
0.2
co
0.2
fe
0.2
mn
0.6
o3(lscfm)初样;
55.s4:将步骤s3制得的lscfm初样转移至管式炉,先用h2/ar混合气(h2含量20%)置换炉内气氛,然后在h2/ar混合气的气氛下(气体流量为100ml/分钟)将管式炉升温至500℃,升温速率0.5℃/分钟,保持500℃条件6小时,使lscfm初样中的ru发生还原,ru纳米粒子原位析出、组装于lscfm样品的表面,最终得到ru@lscfm催化剂。
56.多孔金属氧化物催化剂的制备例2:
57.au@lsc催化剂的制备方法,具体包括以下步骤:
58.s1:称取1.9495g的la(no3)3,0.8465g的sr(no3)2,1.8294g的co(no3)2、3.3979g的haucl4溶入120ml去离子水中搅拌均匀,然后加入7.2966g的柠檬酸铵,柠檬酸铵能够有效络合金属盐离子,得到金属前驱体溶液;
59.s2:称取30ml步骤s1制得的金属前驱体溶液通过5-7次浸渍将溶液完全浸渍于2g球形炭模板的表面,静置24h后加入尿素0.6000g、peg-ppg-peg0.58g、乙醇0.46g、乙二醇0.6207g搅拌均匀,然后将混合物置于烘箱内,在180℃条件下水热合成24h,得到样品;
60.其中,球形炭模板包括线型酚醛树脂和聚乙二醇,线型酚醛树脂和聚乙二醇的质量比为100:17.6;
61.s3:将步骤s2得到的样品置于马弗炉内焙烧10h(900℃,升温速率1℃/min),确保氧化去除球形炭模板剂,得到la
0.6
sr
0.4
coo3(lsc)初样;
62.s4:将步骤s3制得的lsc初样转移至管式炉,先用co/he混合气(co含量5wt%)置换炉内气氛,然后在co/he混合气的气氛下(气体流量为200ml/分钟)将管式炉升温至400℃,升温速率1℃/分钟,保持400℃条件8小时,使lsc初样中的au发生还原,au纳米粒子原位析出、组装于lsc样品的表面,最终得到au@lsc催化剂。
63.多孔金属氧化物催化剂的制备例3:
64.ag@cemno
x
催化剂的制备方法,具体包括以下步骤:
65.s1:称取1.6987g的agno3、3.2613g的ce(no3)3、1.7895g的mn(no3)2溶入90ml去离子水中搅拌均匀,然后加入1.0514g的二乙醇胺,二乙醇胺能够有效络合金属盐离子,得到金属前驱体溶液;
66.s2:称取20ml步骤s1制得的金属前驱体溶液通过5-7次浸渍将溶液完全浸渍于2g的n-甲基吡咯烷模板的表面,静置24h后转移至烘箱内,在110℃条件下真空干燥12小时;
67.s3:将步骤s2得到的样品置于马弗炉内焙烧8h(700℃,升温速率0.5℃/min),完全去除模板剂,得到cemno
x
初样;
68.s4:将步骤s3制得的cemno
x
初样转移至线筒式等离子体反应器内(dbd电极结构,管外径与内径分别为10mm与8mm,床层高度10mm),先用h2/ar混合气(h2含量10wt%)置换炉内气氛,然后在连续通入h2/ar混合气的气氛下(气体流量为100ml/分钟)进行dbd等离子体放电,15khz正弦波交流电源,放电功率15w,连续放电40分钟,使cemno
x
初样中的ag发生还原,ag纳米粒子原位析出、组装于cemno
x
样品的表面,最终得到ag@cemno
x
催化剂。
69.多孔金属氧化物催化剂的对比制备例1:
70.ru@lscfm催化剂的制备方法,具体包括以下步骤:
71.s1:称取2.5994g的la(no3)3,0.4233g的sr(no3)2,0.3659g的co(no3)2,0.4837g的fe(no3)3,1.0737g的mn(no3)2溶入100ml去离子水中搅拌均匀,然后加入6.3042g的柠檬酸,柠檬酸能够有效络合金属盐离子,得到金属前驱体溶液;
72.s2:将步骤s1得到的金属前驱体溶液70℃下搅拌5h,络合充分后,继续在70℃下搅拌约6h得到较为粘稠的溶液;
73.s3:将步骤s2得到的粘稠溶液转移至瓷碗在加热炉上自蔓延燃烧30min,得到灰黑色粉末,然后置于马弗炉内900℃下焙烧6h;得到lscfm初样;
74.s4:将步骤s3得到的lscfm初样与rucl3等体积混合,搅拌均匀后超声10min,然后静置14h,
75.s5:将静置后的样品水洗两次,然后转移至烘箱内,在110℃条件下真空干燥6小时;
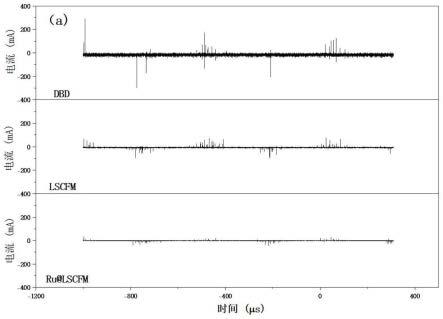
76.s6:将步骤s5得到的样品转移至管式炉,先用h2/ar混合气(h2含量20%)置换炉内气氛,然后在h2/ar混合气的气氛下(气体流量为100ml/分钟)将管式炉升温至500℃,升温速率0.5℃/分钟,保持500℃条件6小时,最终得到负载ru的lscfm催化剂。
77.多孔金属氧化物催化剂的对比制备例2:
78.au@lsc催化剂的制备方法,具体包括以下步骤:
79.s1:称取1.9495g的la(no3)3,0.8465g的sr(no3)2,1.8294g的co(no3)2溶入120ml去离子水中搅拌均匀,然后加入6.3042g的柠檬酸铵,柠檬酸铵能够有效络合金属盐离子,得到金属前驱体溶液;
80.s2:将步骤s1得到的金属前驱体溶液70℃下搅拌5h,络合充分后,继续在70℃下搅拌约6h得到较为粘稠的溶液;
81.s3:将步骤s2得到的粘稠溶液转移至瓷碗在加热炉上自蔓延燃烧30min,得到灰黑色粉末,然后置于马弗炉内900℃下焙烧6h;得到lsc初样;
82.s4:将步骤s3得到的lsc初样与haucl4等体积混合,搅拌均匀后超声10min,然后静置14h,
83.s5:将静置后的样品水洗两次,然后转移至烘箱内,在110℃条件下真空干燥6小时;
84.s6:将步骤s5得到的样品转移至管式炉,先用h2/ar混合气(h2含量20%)置换炉内气氛,然后在h2/ar混合气的气氛下(气体流量为100ml/分钟)将管式炉升温至500℃,升温速率0.5℃/分钟,保持500℃条件6小时,最终得到负载au的lsc催化剂。
85.多孔金属氧化物催化剂的对比制备例3:
86.ag@cemno
x
催化剂的制备方法,具体包括以下步骤:
87.s1:称取3.2613g的ce(no3)3、1.7895g的mn(no3)2溶入90ml去离子水中搅拌均匀,然后加入1.0514g的二乙醇胺,二乙醇胺能够有效络合金属盐离子,得到金属前驱体溶液;
88.s2:将步骤s1得到的金属前驱体溶液70℃下搅拌5h,络合充分后,继续在70℃下搅拌约6h得到较为粘稠的溶液;
89.s3:将步骤s2得到的粘稠溶液转移至瓷碗在加热炉上自蔓延燃烧30min,得到灰黑色粉末,然后置于马弗炉内900℃下焙烧6h,得到cemno
x
初样;
90.s4:将步骤s3得到的lsc初样与agno3等体积混合,搅拌均匀后超声10min,然后静置14h,
91.s5:将静置后的样品水洗两次,然后转移至烘箱内,在110℃条件下真空干燥6小时;
92.s6:将步骤s5得到的样品转移至管式炉,先用h2/ar混合气(h2含量20%)置换炉内气氛,然后在h2/ar混合气的气氛下(气体流量为100ml/分钟)将管式炉升温至500℃,升温速率0.5℃/分钟,保持500℃条件6小时,最终得到负载ag的cemno
x
催化剂。
93.实施例
94.本技术所涉及的等离子体放电反应器主要由阵列式线-线电极,反应室,匹配系统,电源系统,供气系统,气体检测系统,电学诊断系统,光学诊断系统,以及催化剂表征系统组成。
95.本技术所涉及的原位耦合的方法构建等离子体催化系统包括:将催化剂置于等离子体放电反应器中,由电源系统激励产生放电等离子体,将放电等离子体的非平衡特性和催化剂的催化性能结合具有一定的协同作用,形成等离子体-催化耦合系统。
96.实施例1
97.多孔金属氧化物催化剂协同等离子体系的构建方法包括:
98.选用组装筒式dbd放电反应器作为等离子体放电反应器,电源系统优选为5khz交流电源;
99.催化剂采用多孔金属氧化物催化剂的制备例1的方法制备而成的ru@lscfm催化剂。
100.将0.5g的ru@lscfm催化剂加入等离子体放电反应器反应室的等离子体区内,利用电学诊断系统和光学诊断系统对等离子体-催化耦合系统进行在线诊断,利用催化剂表征系统对催化剂进行表征,并研究等离子体与催化剂的协同作用机理。
101.确定等离子体-催化耦合系统的最优构建条件为:dbd放电管外径10mm,内径8mm,放电管外部为ag浆涂覆的地电极(厚度0.5mm),高压电极为直径3mm的不锈钢棒;催化剂为均匀的颗粒,粒径20-40目,系统的床层高度为6mm;
102.实施例2
103.多孔金属氧化物催化剂协同等离子体系的构建方法包括:
104.选用组装平板式混合放电反应器作为等离子体放电反应器,电源系统优选为尖波脉冲电源,输入电压为10kv,脉宽为200ns;
105.催化剂采用多孔金属氧化物催化剂的制备例2的方法制备而成的au@lsc催化剂;
106.将0.5g的au@lsc催化剂加入等离子体放电反应器反应室的等离子体区内,利用电学诊断系统和光学诊断系统对等离子体-催化耦合系统进行在线诊断,利用催化剂表征系统对催化剂进行表征,并研究等离子体与催化剂的协同作用机理。
107.确定等离子体-催化耦合系统的最优构建条件为:平板反应器的长度为10cm,宽度为7cm,板间距为5mm,两不锈钢栅状电极分别置于上下两陶瓷板,栅状电极交错放置,栅状电极面积6cm*6cm,栅条宽2mm,栅条间距1mm,电极厚度为0.5mm;催化剂为均匀的粉体,将催化剂涂敷于上下陶瓷板的栅状电极的栅条空隙内,催化剂涂层厚度为1mm。
108.实施例3:
109.多孔金属氧化物催化剂协同等离子体系的构建方法包括:
110.选用组装气动式滑动弧放电反应器作为等离子体放电反应器,电源系统优选为尖波脉冲电源,输入电压为10kv,脉宽为200ns;
111.催化剂采用多孔金属氧化物催化剂的制备例3的方法制备而成的ag@cemno
x
催化剂;
112.将0.5g的ag@cemno
x
催化剂加入等离子体放电反应器反应室的等离子体区后端,利用电学诊断系统和光学诊断系统对等离子体-催化耦合系统进行在线诊断,利用催化剂表征系统对催化剂进行表征,并研究等离子体与催化剂的协同作用机理。
113.确定等离子体-催化耦合系统的最优构建条件为:滑动弧反应器的结构为筒形,高度为8cm,筒外直径为5cm,筒壁厚为5mm,筒作地极;高压端使用内置纺锤状的不锈钢电极,高压电极与筒壁最小间距处为3mm,引弧在这个位置发生;催化剂堆积为7mm高的床层,紧邻滑动弧区放置;放电反应器的进气为切向进气,流速5m/s。
114.对比例
115.对比例1:和实施例1的区别在于,未加入ru@lscfm催化剂。
116.对比例2:和实施例1的区别在于,催化剂采用多孔金属氧化物催化剂的对比制备例1的方法制备而成。
117.对比例3:和实施例2的区别在于,未加入au@lsc催化剂。
118.对比例4:和实施例2的区别在于,催化剂采用多孔金属氧化物催化剂的对比制备例2的方法制备而成。
119.对比例5:和实施例3的区别在于,未加入ag@cemno
x
催化剂。
120.对比例6:和实施例3的区别在于,催化剂采用多孔金属氧化物催化剂的对比制备例3的方法制备而成。
121.性能检测试验
122.1、将上述实施例1和对比例1-2提供的多孔金属氧化物催化剂协同等离子体系用于典型vocs-甲苯的氧化脱除研究试验,以评价其性能,检测结果见图1和图2。
123.实验条件如下:100ppm甲苯、20vol%o2与79vol%n2组成的气体以100000h-1
的空速单程流经等离子体催化系统。
124.结合实施例1和对比例1-2并结合图1可以看出,从放电特性上看(见图1),在相同输入功率下(10w),单纯dbd放电的电流幅值约为300ma,lscfm催化剂置入放电区可使电流幅值降至约100ma,而ru@lscfm催化剂与dbd耦合放电的电流幅值仅为约50ma,这说明基于dbd放电反应器与ru@lscfm催化剂内置式原位耦合构建的等离子体催化系统能有效调变dbd放电特性,使放电变得更加均匀。
125.结合实施例1和对比例1-2并结合图2可以看出,从反应性能上看(图2),在相同条件下,基于dbd放电反应器与ru@lscfm催化剂内置式原位耦合构建的等离子体催化系统能获得最高的甲苯脱除率(92%),co2选择性(88%)与碳平衡(95%),这可主要归因于该系统的多场高效耦合特性;同样条件下,采用dbd放电与常规方法得到的lscfm催化剂内置式原位耦合所得系统仅能得到77%的甲苯脱除率、53%的co2选择性与85%的碳平衡,而常规dbd放电的甲苯脱除率只有60%,co2选择性为41%,碳平衡为76%。
126.2、将上述实施例2和对比例3-4提供的多孔金属氧化物催化剂协同等离子体系用于炭催化燃烧反应试验,以评价其性能,检测结果见图3和图4。
127.实验条件如下:炭粉填装于平板反应器内,模拟空气(20%o2+80%n2)以20000h-1
的空速单程流经等离子体催化系统。
128.结合实施例2和对比例3-4并结合图3可以看出,从催化剂外观形貌上来看(见图3),au@lsc催化剂较常规lsc催化剂而言,表面含有大量的au纳米粒子,这为等离子体放电在催化剂表面的有效延展提供了点位,同时也为炭催化燃烧反应提供了高活性的表面催化位点。
129.结合实施例2和对比例3-4并结合图4可以看出,从反应性能上看(图4),在相同的放电条件下,经过相同的放电时间处理(1小时),基于混合放电反应器与au@lsc催化剂内置式原位耦合构建的等离子体催化系统能获得最高的炭粉转化率(95%),产物中co2的选择性接近100%,这可主要归因于该系统的多场高效耦合特性;同样条件下,采用混合放电与常规方法得到的lsc催化剂内置式原位耦合所得系统仅能得到71%的炭粉转化率,产物co2的选择性分别是83%;而常规dbd放电的炭粉转化率不足40%,合成气的选择性不足45%。
130.3、将上述实施例3和对比例5-6提供的多孔金属氧化物催化剂协同等离子体系用于ch4与co2干重整反应试验,以评价其性能,检测结果见表1。
131.实验条件如下:50vol%co2与50vol%ch4组成的气体以200000h-1
的空速单程流经
等离子体催化系统。
132.表1:检测结果
[0133][0134]
结合实施例3和对比例5-6并结合表1可以看出,从反应性能上看,在相同的输入功率下(40w),基于滑动弧放电反应器与ag@cemno
x
催化剂耦合构建的等离子体催化系统能获得最高的ch4与co2转化率(98%与95%),产物中合成气的选择性接近100%,反应碳平衡一直保持在99%以上,这可主要归因于该系统的多场高效耦合特性;同样条件下,采用滑动弧放电与常规方法得到的cemno
x
催化剂耦合所得系统仅能得到82%的ch4转化率、75%的co2转化率,产物h2与co的选择性分别是87%和89%,碳平衡约为90%;而常规滑动弧放电的反应物转化率均不足60%,合成气的选择性不足70%,碳平衡为72%。更重要的是,基于ag@cemno
x
催化剂与滑动弧放电构建的等离子体催化系统在干重整反应中连续48小时未出现明显失活现象,如图5所示;滑动弧放电在干重整反应中活性始终随反应进行而下降,如图6所示;由常规cemno
x
催化剂与滑动弧放电构建的等离子体催化系统在干重整反应过程中活性也缓慢下降,其原因主要在于反应副产物在催化剂表面的累积。
[0135]
结合实施例3和对比例5-6并结合图7可以看出,ag@cemno
x
催化剂较常规cemno
x
催化剂而言,表面含有大量的ag纳米粒子为co2与ch4干重整反应提供了高活性的表面催化位点。
[0136]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1