本发明属于有机合成,具体地说,涉及一种用于制备环丙甲酮的催化剂及其制备方法和环丙甲酮的制备方法。
背景技术:
1、环丙甲酮是一种重要的有机合成原料,在诸多化合物的制备中起到引入环丙基结构的作用,而这类结构可以改善产物的脂溶性与代谢速率。例如德国拜耳公司开发了一种三唑硫酮类绿色杀菌剂丙硫菌唑,用于防治小麦病害,可在自然环境中降解无残留。美国默克公司研制了依法韦仑,作为一种非核苷类逆转录酶抑制剂,与其他药物配伍,用于hiv-1感染病人的治疗。此外,环丙甲酮具有张力环结构,可用于制备一系列合成燃料,提高其能量密度、比冲等关键特性,进而提高运载能力。因此,对环丙甲酮工业制备方法的研究具有非常重要的意义。
2、目前环丙甲酮的工业制备的一种方法为采用2-甲基呋喃为原料,经由催化氢化、氯化与成环反应三步制得。催化氢化步骤中采用的pd/c催化剂易被原料中的痕量杂质毒化,导致催化剂的催化活性降低,进而导致工艺周期延长,环丙甲酮的产率偏低;并且氯化与成环步骤中会大量消耗酸碱,产生含盐废水与无实际用途的焦油废液,导致环丙甲酮的产率偏低。
3、环丙甲酮的工业制备的另一种方法为碱金属碘化物如碘化钠、碘化钾等催化2-乙酰基-γ-丁内酯的裂解,一步即可生成产物环丙甲酮。该方法只生成co2副产物,不排放其他有毒有害的三废,具有环境友好性。然而该工艺需要150℃以上的高温条件,使得碘化物催化剂在此条件下易失活,催化活性大大降低,而加入高沸点极性溶剂如n-甲基吡咯烷酮、二甲基丙烯脲或六甲基磷酰三胺等虽然可以有效抑制碘化物催化剂失活,但此时副反应会被加速,从而引入新的难分离杂质,降低产品纯度。
4、中国发明专利cn106554263 a公开了一种新的环丙甲酮的制备方法。仅需催化加氢与酸催化重排两步,即可在较低温度下,以较为可观的产率制得环丙甲酮,同时避免了焦油副产物生成与大量三废排放,但是该方法仍需两步反应以及不同种类的催化剂,延长了反应后处理过程,增加了反应混合物分离难度。
5、有鉴于此特提出本发明。
技术实现思路
1、本发明要解决的技术问题在于克服现有技术的不足,提供一种用于制备环丙甲酮的催化剂及其制备方法和环丙甲酮的制备方法。一方面所述催化剂能够提高催化活性,进而提高环丙甲酮的产率;另一方面所述催化剂的制备方法简单,可批量制备用于工业放大,具有良好的循环复用性;另一方面所述环丙甲酮的制备方法能够避免高压氢气的使用,提高了工艺安全性,缩短了工艺周期,具有产物易分离、三废少、工艺简单的特点,为规模化生成提供了可行方案。
2、
3、为解决上述技术问题,本发明采用技术方案的基本构思是:
4、本发明提供一种用于制备环丙甲酮的催化剂,包括酸性载体和负载于酸性载体表面的贵金属;
5、在催化剂中,相对于100重量份的酸性载体,贵金属的含量为0.1-5重量份。
6、进一步地,在催化剂中,相当于100重量份的酸性载体,贵金属的含量为2-4重量份;
7、优选地,在催化剂中,相当于100重量份的酸性载体,贵金属的含量为4重量份。
8、可以概括为贵金属占酸性载体的质量百分比为0.1%-5%;
9、在上述限定的范围内,反应产率随贵金属负载量的提高而提高。
10、作为最优选的方案,贵金属占酸性载体的质量百分比为4%;
11、当贵金属负载量达到4%后,继续增大贵金属的负载量将会遮蔽酸性载体的酸性位点,阻碍重排反应,导致反应产率降低。因此,在保证产物具有高的产率的前提下,选用贵金属占载体的质量百分比为4%。
12、进一步地,酸性载体选自氧化铝、二氧化钛、二氧化锆、二氧化铪、五氧化二钒、五氧化二铌或五氧化二钽的一种或多种组合;
13、贵金属选自钯、铂、钌和铑中的一种或多种组合;
14、优选地,酸性载体为二氧化钛,贵金属为钯。
15、当贵金属为钯时,产氢效率最高,能够加速环丙甲酮的生成。
16、进一步地,酸性载体二氧化钛为二氧化钛p25,其中,二氧化钛p25购买自赢创-德固赛公司。二氧化钛p25作为载体具有较大的比表面积(约50m2/g),进而增加贵金属的负载量并使其均匀分散,从而显著提高催化性能,使催化剂稳定性高,不易失活,并且该载体的酸性较强,可有效促进重排反应的发生。
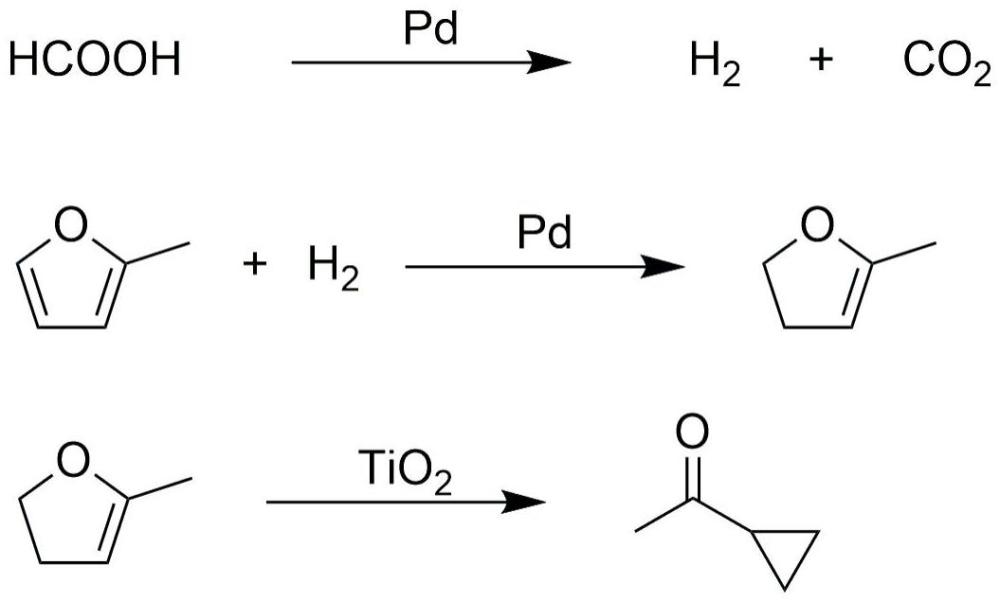
17、进一步地,将0.1-5重量份的贵金属氯化物粉体溶于水中,制得贵金属氯化物的水溶液;
18、将100重量份的酸性载体粉体与贵金属氯化物的水溶液混合,对混合物依次进行干燥、冷却处理,制得干燥的催化剂前驱体;
19、将催化剂前驱体依次进行煅烧、还原、冷却处理,制得催化剂粉体。
20、上述催化剂的制备方法为等体积浸渍-还原法,制备得出的催化剂为粉体,贵金属氯化物可为氯化钯、四氯化铂、三氯化钌和三氯化铑中的一种或多种组合,制备过程中在搅拌下向贵金属氯化物水溶液中加入酸性载体粉体,或者在搅拌下将贵金属氯化物水溶液加入至酸性载体粉体中。
21、加入的贵金属氯化物占载体的质量百分比为0.1%-5%,相当于100重量粉的酸性载体,贵金属的含量为0.1-5重量份。
22、本发明还提供一种应用上述技术方案的催化剂的环丙甲酮的制备方法,
23、2-甲基呋喃和供氢溶剂在催化剂的作用下发生催化氢转移、催化加氢和重排反应,制得到环丙甲酮。
24、在上述方案中,贵金属以单质形式负载于酸性载体上,贵金属起到催化分解供氢溶剂制氢与催化2-甲基呋喃加氢的作用,贵金属首先催化供氢溶剂分解,促使氢气生成,然后催化吸附在表面的2-甲基呋喃加氢,生成2-甲基-4,5-二氢呋喃。酸性载体为2-甲基-4,5-二氢呋喃发生重排反应提供酸性条件,可有效促进重排反应的发生,有效促进2-甲基呋喃生成环丙甲酮。
25、进一步地,供氢溶剂在催化剂中贵金属的作用下发生催化氢转移反应生成氢气和二氧化碳;
26、2-甲基呋喃在催化剂中贵金属的作用下发生催化加氢反应生成2-甲基-4,5-二氢呋喃;
27、2-甲基-4,5-二氢呋喃在催化剂中酸性载体提供的酸性条件下发生重排反应,制得环丙甲酮。
28、进一步地,将2-甲基呋喃、供氢溶剂和催化剂粉体进行混合;
29、将混合物加热至反应温度进行反应,并将反应温度保持至反应结束;
30、将反应结束得到的产物进行处理得到环丙甲酮。
31、进一步地,供氢溶剂选自甲酸、甲酸铵、甲酸肼、乙酸肼、盐酸肼、硫酸肼中的一种或多种组合。
32、在上述方案中,供氢溶剂在贵金属的催化下,分子内的负氢转移至催化剂表面,并与质子结合,原位产生氢气,因此上述供氢溶剂可以作为储氢材料使用。相比于高压氢气,上述供氢溶剂的安定性较好,爆炸极限较窄,且常温下为液体或固体,便于投料操作,增强了工艺安全性。
33、甲酸价格最低,其分解制氢副产物仅为co2,易于去除且不干扰反应,因此优选供氢溶剂为甲酸。
34、进一步地,反应温度为40℃-120℃;
35、供氢溶剂与2-甲基呋喃的摩尔比为1.0:1-1.4:1;
36、酸性催化剂的质量为2-甲基呋喃的质量的1%-10%;
37、优选地,反应温度为50-100℃;
38、供氢溶剂与2-甲基呋喃的摩尔比为1.1:1-1.3:1;
39、酸性催化剂的质量为2-甲基呋喃的质量的5%-8%;
40、进一步优选地,反应温度为60℃;
41、供氢溶剂与2-甲基呋喃的摩尔比为1.1:1;
42、酸性催化剂的质量为2-甲基呋喃的质量的6%。
43、在上述方案中,需要保证供氢试剂过量于环丙甲酮,能够提供足够的氢气,但供氢试剂用量过高会促使反应过程中的中间体继续加氢,生成2-甲基四氢呋喃,降低反应选择性,因此供氢溶剂与2-甲基呋喃的摩尔比为1.0:1-1.4:1;为了进一步提高环丙甲酮的产率,优选的供氢溶剂与2-甲基呋喃的摩尔比为1.1:1。
44、在上述限定的温度下,2-甲基呋喃可顺利转化为环丙甲酮,但是若反应温度过高,将会产生副反应,降低反应选择性;若反应温度过低则反应过慢,降低生产效率;
45、当催化剂的质量与环丙甲酮的质量在上述限定范围内时,催化剂具有良好的催化效果,后处理方便,生产效率高;但是催化剂用量大,产物选择性下降;催化剂用量小,延长了反应时间,降低生产效率。
46、进一步地,制备环丙甲酮的反应时间为1h-7h;
47、优选地,反应时间为4h-5h,
48、进一步优选地,反应时间为5h。
49、在上述方案中,反应时间可以理解为反应物与催化剂接触的时间。在上述限定的反应时间范围下,可使2-甲基呋喃与催化剂既能充分接触,催化反应正常进行,又不会产生过度反应,保证原料的利用率和产物的产率,提高产物的选择性。若反应时间过短,原料无法及时转化,产率过低;若反应时间过长则容易生成副产物,使产物的选择性降低。
50、进一步的,制备环丙甲酮的反应压力为0.1mpa-2mpa,优选为0.1mpa-1mpa。因2-甲基呋喃沸点较低,提高反应温度后,体系压力增大,对反应器内壁材料的有一定的耐压要求,因此,反应压力限定在0.1mpa-2mpa范围内;若压力过高,则承压要求更加苛刻,造成安全隐患。
51、进一步的,2-甲基呋喃的纯度不低于98.5%,供氢试剂的纯度不低于99%。
52、在上述方案中,原料2-甲基呋喃的纯度较高,有利于反应顺利进行;纯度过低则会使催化剂中钯中毒,导致反应停止;
53、2-甲基呋喃与供氢试剂均为工业原料,可便利地购买。
54、具体地,向反应釜内装填催化剂、供氢溶剂和2-甲基呋喃,将反应釜内进行氩气置换后,调整温度至反应温度,反应釜恒温后,保温至反应结束,将反应结束得到的产物进行处理得到环丙甲酮。过程中,通过搅拌使2-甲基呋喃与供氢试剂充分接触催化剂,生成环丙甲酮。
55、进一步地,将反应结束得到的产物依次进行过滤,得到产物滤液和催化剂滤饼;
56、将催化剂滤饼进行回收重复利用;
57、将产物滤液进行蒸馏,收集常压下、设定温度下的馏分,收集的馏分为环丙甲酮。
58、在上述方案中,将反应结束得到的产物进行处理的步骤包括过滤、蒸馏,具体为:使用疏水聚四氟乙烯滤膜,其中,滤膜的孔径选用450nm,对反应结束得到的产物进行过滤,回收催化剂用于下一次反应,所得滤液使用垂刺精馏柱进行蒸馏,收集常压下114℃±1℃馏分,馏分即为环丙甲酮。
59、按照本发明的方法制备产物环丙甲酮的产率高达98%,纯度高达99.8%。
60、采用上述技术方案后,本发明与现有技术相比具有以下有益效果:
61、1、本发明的催化剂能够提高催化活性,进而能够提高环丙甲酮的产率;
62、2、本发明的催化剂的制备方法简单,可批量制备用于工业放大,具有良好的循环复用性。
63、3、本发明的环丙甲酮的制备方法能够避免高压氢气的使用,价格低廉,提高了工艺安全性;以及,
64、本发明的环丙甲酮的制备方法采用一锅法的工艺,在提高产物的产率的基础上,能够缩短工艺周期,具有产物分离少、三废少、工艺简单的特点,有利于工艺放大,为后续的产品规模化生成奠定了基础。
65、下面对本发明的具体实施方式作进一步详细的描述。