一种电极及其制备方法、电解装置与流程
本发明涉及电催化技术领域,尤其涉及一种电极的制备方法、电极及电解装置。
背景技术:
电催化还原技术的主要优点是:无需消耗化学试剂、多功能性、高能源效率、环境兼容性以及容易控制等。电催化还原技术中决定电催化活性高低的关键部件是工作电极,它是电化学催化系统的“心脏”。如在co2的电催化还原中,电极材料对提高co2催化转化效率至关重要。
在现有电极材料中,具有微孔结构的碳基材料拥有较高的比表面积,但是对于电催化性能并没有明显提高;而且碳基材料存在制备步骤繁琐、工艺过程复杂、制备条件苛刻以及成本较高等问题,限制了其在电催化领域(尤其是二氧化碳电催化领域)的进一步应用。而铜基材料由于具有相对较好的电催化性能,特别是其具有廉价易得、工作温度范围宽、生产工艺成熟、对环境友好等特点,因此,目前制作电极时主要采用铜基材料。
但是基于单纯铜基材料的电催化材料的电催化性能并不理想。为提高基于铜基材料的电催化材料的电催化性能,目前针对铜基材料的研究方向主要集中在两方面:
一方面,通过提高铜基材料的比表面积,增加电催化反应中电极材料与电解液的接触面积,来提高其电催化性能。另一方面,利用贵金属纳米颗粒对铜基材料进行修饰。由于电极用于电催化反应中时,电催化反应产物吸附在电极表面会引起电极表面的污染和钝化,或因其长期使用,电极表面本身发生的物理和化学变化,会导致其催化活性、催化重现性和稳定性大大降低,利用贵金属纳米颗粒对铜基材料进行修饰可有效降低电极表面的污染和钝化,减小其表面的物理和化学变化。同时,贵金属纳米颗粒和铜基材料所产生的协同作用可进一步提高电极的电催化性能。
利用贵金属纳米颗粒对铜基材料进行修饰,可得到具有高比表面积的多层次结构的材料。但是这样的材料不具备自支撑特性,所以在制作电极的过程中需要添加粘结剂和活性炭等辅助材料。这种制备方式对形成后的电极比表面有所影响,会降低电极中的活性材料与电解液的有效接触面积,并且还会导致形成的电极材料具有较大的接触电阻,使得采用现有技术的制备方法获得的上述电极应用于电催化反应中时,限制了催化反应中目标产物的法拉第效率的进一步提高。
技术实现要素:
有鉴于此,为解决现有技术的问题,为克服上述现有技术中的缺陷,本发明的实施例提供一种电极及其制备方法、电解装置,采用该制备方法获得的电极具有自支撑性能和较大的比表面积,应用于电催化反应中可提高催化反应中目标产物的法拉第效率。
为达到上述目的,本发明的实施例采用如下技术方案:
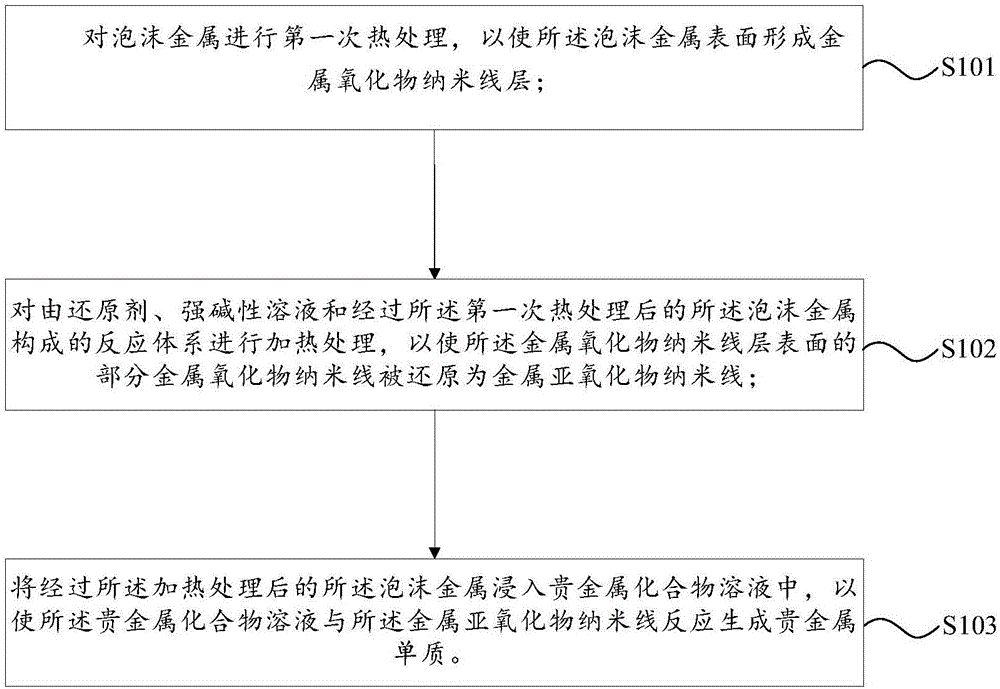
一方面、本发明实施例提供了一种电极的制备方法,所述制备方法包括:对泡沫金属进行第一次热处理,以使所述泡沫金属表面形成金属氧化物纳米线层;对由还原剂、强碱性溶液和经过所述第一次热处理后的所述泡沫金属构成的反应体系进行加热处理,以使所述金属氧化物纳米线层表面的部分金属氧化物纳米线被还原为金属亚氧化物纳米线;将经过所述加热处理后的所述泡沫金属浸入贵金属化合物溶液中,以使所述贵金属化合物溶液与所述金属亚氧化物纳米线反应生成贵金属单质。
可选的,所述制备方法还包括:在惰性气氛下,对形成有所述贵金属单质的所述泡沫金属进行第二次热处理,以使残留于表面的所述贵金属的化合物溶液中的化合物分解形成贵金属单质。
可选的,所述第二次热处理的温度为700-1000℃,时间为5-20min。
可选的,所述第一次热处理的温度为400-700℃,时间为0.5-3h。
可选的,所述加热处理的温度为160-200℃,时间为3-12h。
可选的,所述将经过所述加热处理后的所述泡沫金属浸入贵金属化合物溶液中的时间为1-6h。
可选的,所述泡沫金属包括:泡沫铜或泡沫镍。
可选的,所述贵金属的化合物溶液包括:金的化合物溶液或铂的化合物溶液。
另一方面、本发明实施例还提供了一种电极,包括:由泡沫金属构成的支撑层;所述电极还包括:形成在所述支撑层表面的金属和氧的化合物纳米线层;形成在所述金属和氧的化合物纳米线层表面的贵金属单质。
可选的,所述金属和氧的化合物纳米线层包括:
金属氧化物纳米线层;或者,金属氧化物纳米线层和形成在所述金属氧化物纳米线层表面的金属亚氧化物纳米线层。
可选的,所述泡沫金属包括:泡沫铜或泡沫镍。
可选的,所述贵金属单质包括:金或铂。
再一方面、本发明实施例还提供了一种电解装置,包括上述所述的电极。
基于此,本发明实施例提供的上述电极的制备方法,以泡沫金属作为集流体和支撑体,通过采用高温预氧化、水热反应(示例的可以为溶剂热反应)以及氧化还原反应相结合的方式,得到一种三元复合材料,该三元复合材料包括泡沫金属、在泡沫金属逐层形成的金属和氧的化合物纳米线层、贵金属单质。其中,由于泡沫金属本身具有一定的自支撑特性,应用到电催化反应中时无需额外设置集流体或支撑体,其本身即可作为集流体和支撑体;并且,由于泡沫金属内部分布着大量孔洞或孔隙结构,具有较高的比表面积,不仅能够加速电化学催化过程中的电子收集和转移速率,还能够提高电极材料与电解液的接触面积和增加电解液的扩散速率,为电催化过程提供更多的活性位点,从而可提高电极的催化效率。而金属和氧的化合物纳米线层是利用预氧化反应自然生长在泡沫金属表面的,无需额外添加粘结剂等辅助材料,不会影响泡沫金属较大的比表面积。形成在上述复合材料表面的贵金属单质能够对泡沫金属的催化性能进行修饰,使得二者之间发生协同作用,进一步提高获得的上述复合电极应用于电催化反应中的目标产物的法拉第效率,即实际生成物与理论生成物的百分比。
此外,本发明实施例提供的上述电极的制备方法,相对于现有的制备方法,制备工艺简单、流程较短、设备依赖性较低,更适用于工业化大规模生产。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例提供的一种电极的制备方法流程示意图;
图2为本发明实施例提供的一种电极的扫描电镜(scanningelectronicmicroscopy,简称为sem)照片;
图3为本发明实施例提供的一种电极与泡沫铜的x射线衍射图谱(x-raydiffraction,简称为xrd);
图4为本发明实施例提供的一种电极应用于电催化反应中时在不同时间点的电催化效率。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。应当理解的是,本发明所有原料,对其来源没有特别限制,在市场上购买或按照本领域技术人员熟知的常规方法制备即可,涉及到的溶液,除特别说明外,溶剂均为离子水。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明实施例提供了一种电极的制备方法,如图1所示,该制备方法包括:
s101、对泡沫金属进行第一次热处理,以使泡沫金属表面形成金属氧化物纳米线层;
s102、对由还原剂、强碱性溶液和经过第一次热处理后的泡沫金属构成的反应体系进行加热处理,以使金属氧化物纳米线层表面的部分金属氧化物纳米线被还原为金属亚氧化物纳米线;
s103、将经过加热处理后的泡沫金属浸入贵金属化合物溶液中,以使贵金属化合物溶液与金属亚氧化物纳米线反应生成贵金属单质。
需要说明的是,第一、在上述s101中,泡沫金属是指含有泡沫气孔的特征金属材料,具有自支撑特性,比表面积大等特性。
可以理解的是,由于上述s101是要使得对泡沫金属进行第一次热处理后,在泡沫金属表面形成金属氧化物纳米线层。因此,上述第一次热处理是在包括有氧气的气氛(示例的可以为空气)下进行的,即对泡沫金属进行高温预氧化处理。
上述s101中金属氧化物纳米线层形成原理为:
在高温与氧气条件下,泡沫金属内部的金属离子通过晶界扩散到表面后受到高温的作用会继续沿着泡沫金属表层晶粒之间的晶界扩散,即在表层晶界处形核,与氧气发生反应生成金属氧化物。内部的金属离子不断沿着形核后的晶粒之间的晶界继续扩散,从而使得金属氧化物沿着晶界生长成纳米线,这些纳米线大量地形成在泡沫金属表面,从而构成了金属氧化物纳米线层,形成了泡沫金属/金属氧化物纳米线层的第一级复合结构,以下表示为m/mo,其中“m”表示泡沫金属的金属元素。
第二、在上述s102中,对由还原剂、强碱性溶液和经过第一次热处理后的泡沫金属构成的反应体系进行加热处理,溶液的量需完全浸没泡沫金属。
在此过程中,还原剂具体为弱还原剂,在强碱性溶液中能够使泡沫金属表面的部分金属氧化物纳米线被还原得到金属亚氧化物纳米线,从而形成了泡沫金属/金属氧化物纳米线层/金属亚氧化物纳米线的第二级复合结构。
上述结构示例的可以表示为m/mxo,其中,下标记“x”表示金属氧化物和金属亚氧化物中金属元素与氧元素的整体配比。
可以理解的是,如果通过强还原剂将金属氧化物纳米线全部还原,容易导致泡沫金属表面的金属氧化物纳米线的形貌遭到破坏,使得泡沫金属表面的金属氧化物纳米线层坍塌,难以形成上述的泡沫金属/金属氧化物纳米线层/金属亚氧化物纳米线的复合结构。
因此,上述s102中通过弱还原剂将金属氧化物纳米线部分还原为金属亚氧化物纳米线。
这里,上述的强碱性溶液通常是指该溶液能使特定指示剂变色的物质(如使紫色石蕊溶液变蓝,使无色酚酞溶液变红等),在标准情况下(浓度为0.1mol/l)),ph值大于12。该强碱性溶液中的强碱在水溶液中能够全部电离,且电离出的阴离子全部是氢氧根离子,与酸反应形成盐和水。
第三、在上述s103中,具有还原性的金属亚氧化物与贵金属化合物溶液中具有氧化性的贵金属离子会发生氧化还原反应,生成贵金属单质。
可以理解的是,当所有的金属亚氧化物纳米线与贵金属离子反应时,则生成的贵金属单质附着于金属氧化物纳米线的表面,从而形成了泡沫金属/金属氧化物纳米线层/贵金属的一种第三级复合结构。
上述结构示例的可以表示为m/mxo/y,其中,y表示贵金属元素;在此情况下,mxo仅表示金属氧化物。
当部分金属亚氧化物纳米线与贵金属离子反应时,则生成的贵金属单质附着于金属氧化物纳米线表面、以及形成在金属氧化物纳米线层表面的金属亚氧化物纳米线表面,从而形成了泡沫金属/金属氧化物纳米线层和金属亚氧化物纳米线/贵金属的另一种第三级复合结构。
上述结构示例的可以表示为m/mxo/y,其中,y表示贵金属元素;在此情况下,mxo表示金属氧化物和金属亚氧化物。
基于此,本发明实施例提供的上述电极的制备方法,以泡沫金属作为集流体和支撑体,通过采用高温预氧化、水热反应(示例的可以为溶剂热反应)以及氧化还原反应相结合的方式,得到一种三元复合材料,该三元复合材料包括泡沫金属、在泡沫金属逐层形成的金属和氧的化合物纳米线层、贵金属单质,示例的可以表示为m/mxo/y。其中,由于泡沫金属本身具有一定的自支撑特性,应用到电催化反应中时无需额外设置集流体或支撑体,其本身即可作为集流体和支撑体;并且,由于泡沫金属内部分布着大量孔洞或孔隙结构,具有较高的比表面积,不仅能够加速电化学催化过程中的电子收集和转移速率,还能够提高电极材料与电解液的接触面积和增加电解液的扩散速率,为电催化过程提供更多的活性位点,从而可提高电极的催化效率。而金属和氧的化合物纳米线层是利用预氧化反应自然生长在泡沫金属表面的,无需额外添加粘结剂等辅助材料,不会影响泡沫金属较大的比表面积。形成在上述复合材料表面的贵金属单质能够对泡沫金属的催化性能进行修饰,使得二者之间发生协同作用,进一步提高获得的上述复合电极应用于电催化反应中的目标产物的法拉第效率,即实际生成物与理论生成物的百分比。
此外,本发明实施例提供的上述电极的制备方法,相对于现有的制备方法,制备工艺简单、流程较短、设备依赖性较低,更适用于工业化大规模生产。
为了提高s101中泡沫金属表面形成金属氧化物纳米线层的程度,在上述s101之前,本发明实施例提供的上述电极的制备方法还包括以下步骤:
对泡沫金属进行预先清理,以去除表面杂质和氧化物层。
示例的,可将泡沫金属浸入浓度小于2mol/l的稀硫酸或稀盐酸溶液中利用稀酸的腐蚀性去除泡沫金属表面杂质和氧化物层。
并且,上述预先清理的过程还可利用超声振荡进行辅助清理,以加快清洗过程和/或提高清洗程度。超声振荡的时间示例的可以为5min。
进一步的,在上述s102之前,本发明实施例提供的上述电极的制备方法还包括以下步骤:
对经过s101获得的泡沫金属/金属氧化物纳米线层依次进行清洗、烘干处理。
示例的,可以对上述产物先用去离子水清洗,再在60-80℃条件下烘干10h,以避免经过前述s101形成的金属亚氧化物再次被氧化,烘干需在真空条件下进行。
进一步的,考虑到贵金属化合物溶液中的贵金属化合物有可能没有完全反应,一些贵金属化合物会附着在三元复合材料表面。因此,为了提高经过上述s101~s103制备出的三元复合材料表面负载的贵金属单质的含量,并提高对贵金属化合物溶液的利用率,在上述步骤s103之后,本发明实施例提供的上述制备方法还包括以下步骤:
在惰性气氛下,对形成有贵金属单质的泡沫金属进行第二次热处理,以使残留于表面的贵金属的化合物溶液中的化合物分解形成贵金属单质。
其中,示例的,上述第二次热处理的温度为700-1000℃,时间为5-20min。
需要说明的是,为了防止泡沫金属和贵金属被氧化而使形成的三元复合材料变脆,破坏其自支撑骨架和纳米线层,所以上述第二次热处理在惰性气氛下进行。
惰性气氛的选择可沿用相关技术,本发明实施例对此不做赘述。
为了进一步说明本发明实施例提供的上述制备方法,下面对上述各步骤中涉及的溶液浓度、反应温度、反应时间等反应条件做具体说明:
在上述s101中,第一次热处理的温度示例的可以为400-700℃,时间为0.5-3h。
在该温度和反应时间范围内,一方面能够保证获得的材料仍然保留有泡沫金属的自支撑骨架,不会使得泡沫金属发生完全氧化而使材料变脆,另一方面还可使得形成的金属氧化物保持均匀的纳米线形貌。
泡沫金属可以是泡沫铜或泡沫镍。由于泡沫铜比泡沫镍的制备成本低,导电性能更好,通常选择泡沫铜作原料制备电极。
在上述s102中,加热处理的温度示例的可以为160-200℃,时间为3-12h;
还原剂示例的可以为葡萄糖、尿素、三聚氰胺、双氰胺中的至少一种,浓度为10-50mg/ml;
强碱性溶液示例的可以为氢氧化钾溶液和/或氢氧化钠溶液,浓度小于200mg/ml。
在上述s103中,将经过加热处理后的泡沫金属浸入贵金属化合物溶液中,以使贵金属化合物溶液与金属亚氧化物纳米线反应生成贵金属单质,由于金属亚离子具有较强的还原性,贵金属离子具有很强的氧化性,所以该过程在室温条件下即可进行,时间为1-6h。
这里,所述“室温”,也称为常温或者一般温度。通常来说,室温有3种范围的定义,即:⑴、23℃±2℃;⑵、25℃±5℃;⑶、20℃±5℃。
s103中的贵金属的化合物溶液可以是金的化合物溶液或铂的化合物溶液,浓度示例的可以为1-25mmol/l。
其中,金的化合物溶液包括氯金酸溶液、aucl溶液、aucl3溶液中的至少一种。
在上述基础上,本发明实施例还提供了一种采用上述制备方法获得的电极,该电极包括泡沫金属;该电极还包括:形成在泡沫金属表面的金属和氧的化合物纳米线层;形成在金属和氧的化合物纳米线层表面的贵金属单质。
其中,上述金属和氧的化合物纳米线层示例的可以包括:金属氧化物纳米线层;或者,金属氧化物纳米线层和形成在所述金属氧化物纳米线层表面的金属亚氧化物纳米线层。
该泡沫金属可以为泡沫铜或泡沫镍;该贵金属单质可以为金或铂。
当泡沫金属为泡沫铜(cufoam),贵金属单质为金(au)时,制备的电极是由铜、氧、金这三元构成的复合材料,该三元复合材料的结构具体为:泡沫铜表面形成有铜和氧的化合物纳米线层;纳米线层表面形成有金单质。
这里,铜和氧的化合物纳米线层示例的可以是氧化铜纳米线层,或者,是氧化铜纳米线层和形成在氧化铜纳米线层表面的氧化亚铜纳米线层。
上述三元复合材料可以表示为cufoam/cuxo/au。
在上述基础上,本发明实施例还提供了一种电解装置,包括上述电极。
示例的,该电解装置(例如电解池)可以为h型结构。在h型结构的电解装置中,采用阳离子交换膜作为隔膜将电解装置分隔成阴极室和阳极室,二氧化碳气体可从阴极室溶液底部通入,电解液为0.1m碳酸氢钠水溶液。
为了进一步说明本发明,以下将以泡沫铜为原料结合具体实施例对本发明提供的一种电极的制备方法、电极及电解装置进行详细描述,以下实施例中所用的试剂,在市场上购买或按照本领域技术人员熟知的常规方法制备即可,涉及到的溶液,除特别说明外,溶剂均为离子水,但是应当理解,这些实施例是在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制,本发明的保护范围也不限于下述的实施例。
实施例1
步骤1:对泡沫铜表面进行预先清理,去除表面杂质和氧化物层;
示例的,可以将泡沫铜浸渍在1m(即1mol/l)稀硫酸中,超声处理5min,然后用去离子水清洗晾干。
此处需要说明的是,为了制备出大小合适的电极,该泡沫铜尺寸可以为1.5cm*3cm。
步骤2:将上述泡沫铜置于加热装置中,进行第一次热处理,处理温度为550℃,处理时间为3h;
示例的,可以将上述泡沫铜置于管式炉中,在与空气连通的条件下进行热处理。
步骤3:在反应容器中,加入葡萄糖溶液和强碱溶液,葡萄糖溶液浓度为50mg/ml,强碱溶液浓度为100mg/ml,混合均匀后,将步骤2制备的样品放入上述溶液中,进行加热处理,使该反应体系进行水热反应,处理温度为180℃,处理时间为3h;
示例的,可以在20ml聚四氟芯中加入葡萄糖、固态的氢氧化钾和12~18ml的去离子水,使上述各物料进行水热反应,葡萄糖的浓度为50mg/ml,氢氧化钾溶液的浓度为100mg/ml,混合均匀后,将步骤2制备的样品放入上述溶液中,进行加热处理,处理温度为180℃,处理时间为3h。
此处需要说明的是,在进行水热反应时,为了使得密封的反应容器(例如为聚四氟芯)中留有一定的空间余量,反应体系通常填充在反应容器容积的3/4处。因此,在加入葡萄糖和固态的氢氧化钾时可以根据其各自的浓度计算出具体需加入量,此处不再做具体计算。
步骤4:对步骤3所制备得到的样品进行清洗和烘干;
示例的,采用去离子水对步骤3所制备得到的样品进行清洗,然后置于80℃烘箱进行烘干。
步骤5:将步骤4中所得样品与氯金酸溶液反应1h,其中氯金酸溶液浓度为10mmol/l;
步骤6:采用去离子水对步骤5中获得的样品进行清洗,然后取出置于80℃烘箱烘干;
步骤7:将步骤6中得到的样品置于加热装置中,进行第二次热处理,处理温度为900℃,处理时间5min。
示例的,可以将步骤6中得到的样品置于管式炉中,进行第二次热处理,处理温度保持在900℃,处理时间持续5min,为防止样品被氧化变脆,破坏其自支撑结构,该反应过程需要在惰性气氛中进行。
本实施例制备的电极材料,具有高的比表面积,丰富的、互相连通的多级孔结构,从而使得电极材料在电催化转化过程中具有选择性和稳定性。
下面通过实验测该电极材料在co2电催化过程中的法拉第效率,该实验采用三电极体系,工作电极为实施例1所制备的电极,参比电极和对电极分别采用饱和甘汞电极和铂丝电极。测得该电极材料在co2电催化过程中,在10ma/cm2的电流密度下,co2转化为co的法拉第效率可达67%。
实施例2:
步骤1:对泡沫铜表面进行预先清理,去除表面杂质和氧化物层;
步骤2:对上述泡沫铜置于加热装置中,进行第一次热处理,处理温度为400℃,处理时间为1h;
步骤3:在反应容器中,加入葡萄糖溶液和强碱溶液,葡萄糖溶液浓度为60mg/ml,强碱溶液浓度为100mg/ml,混合均匀后,将步骤2制备的样品放入上述溶液中,进行加热处理,使该反应体系进行水热反应,处理温度为160℃,处理时间为3h;
步骤4:对步骤3所制备得到的样品进行清洗和烘干;
步骤5:将步骤4中所得样品与氯金酸反应1h,其中氯金酸浓度为10mmol/l;
步骤6:将步骤5中获得的样品,用去离子水清洗,取出置于80℃烘箱烘干;
步骤7:将步骤6中得到的样品置于加热装置中,进行第二次热处理,处理温度为1000℃,处理时间5min。
此实例所制的电极在co2电催化过程中,在10ma/cm2的电流密度下,co2转化为co的法拉第效率可达40%。
实施例3
步骤1:对泡沫铜表面进行预先清理,去除表面杂质和氧化物层;
步骤2:对上述泡沫铜置于加热装置中,进行第一次热处理,处理温度为700℃,处理时间为2h;
步骤3:在反应容器中,加入葡萄糖溶液和强碱溶液,葡萄糖溶液浓度为10mg/ml,强碱溶液浓度为100mg/ml,混合均匀后,将步骤2制备的样品放入上述溶液中,进行加热处理,使该反应体系进行水热反应,处理温度为190℃,处理时间为2h;
步骤4:对步骤3所制备得到的样品进行清洗和烘干;
步骤5:将步骤4中所得样品与氯金酸反应1h,其中氯金酸浓度为25mmol/l;
步骤6:将步骤5中获得的样品,用去离子水清洗,取出置于80℃烘箱烘干;
步骤7:将步骤6中得到的样品置于加热装置中,进行第二次热处理,处理温度为800℃,处理时间10min。
此实例所制得的电极材料在co2电催化过程中,在10ma/cm2的电流密度下,co2转化为co的法拉第效率可达50%。
以实施例1为例,通过上述制备过程合成的电极的形貌如图2所示,由图2中可以看出,电极材料表面有丰富的纳米线构成的纳米线层,具有高的比表面积,具有丰富的、互相连通的多级孔隙结构。
图3为泡沫铜(cufoam)与通过上述实施例1合成的电极的物相(xrd)图谱对比结果,由图3中的曲线对比可知,所得电极材料为三元复合材料。
通过实验测试实施例1制得的电极材料在不同时间点的电催化效率,可得到如图4所示的曲线,由图可知,该电极材料应用于电催化反应中,不同的时间点都能达到相应的效率。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种用于非对称电容电池的电容电极制备方法与流程
- 一种超级电容器的电极材料及其制备方法与流程
- 一种超级电容器电极材料PANI/CeO2/Ni(OH)2多级微球及其制备方法与流程
- 流动体系下再生水管道系统腐蚀电化学测试装置与方法与流程
- 电化学电容器用电极材料、电化学电容器用电极涂布液、电化学电容器用电极及电化学电容器的制造方法与工艺
- 一种柔性超级电容器复合电极材料的制备方法与制造工艺
- 用于电化学存储器的分离器、制造电极材料的方法以及电化学储能器与制造工艺
- 电化学组装聚吡咯/二氧化锰复合物改性电极及其制备方法和应用与制造工艺
- 一种超级电容器极片、超级电容器及极片处理方法与制造工艺
- 一种基于双极电极阵列的可视化电化学发光传感器检测葡萄糖的方法与制造工艺