一种医用镁合金表面羟基磷灰石和氧化石墨烯复合生物涂层的电沉积制备方法与流程
本发明涉及生物医用复合材料制备技术领域,更具体的是,本发明涉及一种医用镁合金表面羟基磷灰石和氧化石墨烯复合生物涂层的电沉积制备方法。
背景技术:
近年来,镁及镁合金已经成为医用植入金属材料研究的热点。与现有骨植入生物材料相比,镁合金具有良好的力学性能,镁合金的密度及弹性模量,与人体骨组织接近,可以有效减小应力遮挡效应;此外,镁合金具有良好的生物相容性及生物活性。更重要的是,镁合金在体内可以被完全降解吸收,作为医用植入材料勿需二次手术取出。然而,镁合金在生理环境下的降解过快,极大的限制了其在临床方面的应用。涂层技术已经被认为是提高镁合金耐腐蚀性能的有效手段。传统镁合金的表面涂层单纯地注重于解决镁合金的耐蚀性,而对于植入体内的医用镁合金来说,还要求其对人体无害并具有较好的生物相容性。
在众多涂层材料中,羟基磷灰石(ha)成为生物医用植入材料中理想的涂层材料。羟基磷灰石是天然骨的主要无机成分,在骨质中约占60wt.%。羟基磷灰石具有良好的生物活性,植入人体后能在短时间内与人体的软硬组织紧密结合。但是,羟基磷灰石的力学性能较差,表现为脆性大、耐磨性不好,目前针对羟基磷灰石的研究,重点是通过与其它材料的复合,来改善其生物力学性能。
氧化石墨烯(go)不但含有大量丰富的含氧官能团,而且具有良好的生物相容性和机械性能。氧化石墨烯能促进羟基磷灰石在模拟体液中的生长从而形成氧化石墨烯/羟基磷灰石复合材料,这种go/ha复合材料能够使干细胞增殖并加速其分化,从而成功的用于骨再生治疗。此外,有研究表明,氧化石墨烯还具有抗菌性能,能够防止植入诱导期间的感染。
含有氧化石墨烯纳米复合材料的制备技术国内外已有不少文献报道。文献报道:壳聚糖/氧化石墨烯/羟基磷灰石复合材料的制备(f.mohandes等rscadv.,2014,4,25993);氧化石墨烯/羟基磷灰石复合材料的制备(guangyaoxiong等materialscharacterization107(2015)419–425)复合支架。以上报道存在以下缺陷:合成羟基磷灰石纳米颗粒后加入前驱液中,致使纳米颗粒不能很好的均匀分散在支架上。
cn102569749a的专利公开了一种石墨烯-羟基磷灰石纳米复合材料及其制备方法,采用氨基酸水热法制备石墨烯-羟基磷灰石纳米复合材料,该发明专利制备条件苛刻,对设备要求高,能耗也高。cn104415399a公开了一种湿化学法制备羟基磷灰石-石墨烯纳米复合粉末的方法,采用氨水调节混合液在碱性条件下得到羟基磷灰石-石墨烯前驱体浆料,然后将前驱体浆料熟化后,形成复合材料,该发明的制备条件虽然简单但是在碱性条件下复合形成,而人体体液的ph为7.5左右,难以保证此复合材料在人体中的机械性能和生物相容性。
技术实现要素:
本发明设计开发了一种医用镁合金表面羟基磷灰石和氧化石墨烯复合生物涂层的电沉积制备方法,在纯镁或者镁合金表面原位电沉积有羟基磷灰石和氧化石墨烯复合生物涂层,并优化了氧化石墨烯的含量,电解质的ph和温度以及沉积电压和沉积时间,使得纯镁或者镁合金表面的生物涂层致密均匀,提高生物涂层的稳定性。
本发明提供的技术方案为:
一种医用镁合金表面羟基磷灰石和氧化石墨烯复合生物涂层的电沉积制备方法,包括如下步骤:
步骤1:将羟基化的纯镁或镁合金在水解的氨丙基三乙氧基硅烷溶液中且室温下浸渍5~60分钟,然后在60~120℃下,加热30~60分钟,得到硅烷化的纯镁或者镁合金;
步骤2:以硅烷化的纯镁或镁合金为阴极,石墨片为阳极,置于氧化石墨烯电解液中进行电沉积,得到具有羟基磷灰石和氧化石墨烯复合涂层的纯镁或者镁合金;
其中,所述氧化石墨烯电解液的ph为4~5,所述氧化石墨烯电解液的温度为55~65℃,所述电沉积的沉积电压为10~15v,沉积时为间0~60min;
其中,所述氧化石墨烯电解液的配置包括:
将0.02~0.05mol/l的ca(no3)2,0.01~0.03mol/l的nh4h2po4,2~6ml/l的h2o2,0.05~100μg/ml的氧化石墨烯加入去离子水中,配置成混合溶液。
优选的是,所述羟基化的纯镁或镁合金的制备包括:
将纯镁或镁合金浸入3~6mol/lnaoh溶液中,浸泡30~120分钟,去离子水清洗后干燥。
优选的是,制备所述羟基化的纯镁或镁合金之前,将纯镁或者镁合金进行预处理:
将纯镁或镁合金切割为20mm×15mm×5mm的长方体;
依次用400#、600#、1000#、1500#、2000#的水磨砂纸进行打磨;
丙酮超声清洗后,去离子水超声清洗5~10分钟,储存于无水乙醇中。
优选的是,所述氨丙基三乙氧基硅烷溶液含有的组分及其体积百分比为:硅烷:5%,乙醇:90%,去离子水:5%。
优选的是,在电沉积过程中,始终在磁力搅拌下进行沉积。
优选的是,电沉积结束后,取出表面沉积有羟基磷灰石和氧化石墨烯复合生物涂层的纯镁或镁合金,自然晾干。
优选的是,配置所述氧化石墨烯电解液时,将所述混合溶液经过超声处理,得到稳定存在的悬浮溶液。
优选的是,所述电沉积的沉积电压为10v,沉积时间为30min。
优选的是,所述氧化石墨烯电解液的ph为4.5,温度为60℃。
优选的是,所述氧化石墨烯电解液的配置包括:
将0.042mol/l的ca(no3)2,0.025mol/l的nh4h2po4,6ml/l的h2o2,100μg/ml的氧化石墨烯加入去离子水中,配置成混合溶液,并经过超声处理,得到稳定存在的悬浮溶液。
本发明所述的有益效果:
本发明克服了单一涂层的不足,提供了一种在纯镁或镁合金表面电沉积制备复合生物涂层的方法,在纯镁或者镁合金表面原位电沉积有羟基磷灰石和氧化石墨烯复合生物涂层,并优化了氧化石墨烯的含量,电解质的ph和温度以及沉积电压和沉积时间,使得纯镁或者镁合金表面的生物涂层致密均匀,提高生物涂层的稳定性,并且该制备方法简单、效率高、易操作、适合大面积生产推广;
(1)单纯的羟基磷灰石涂层,其结构为粗大的片状结构,且表面起伏不平,均匀性较差;加入氧化石墨烯后,氧化石墨烯为羟基磷灰石提供了更多的形核位置,使羟基磷灰石的结构由粗大的片状结构变为细小的纳米针叶状结构,且氧化石墨烯均匀的分散在涂层中,整体涂层更加均匀致密,有效的提高了涂层与基体的结合强度;这种细小的纳米针叶状结构,可以促进细胞的黏附,有效的改善了镁合金基体的生物相容性。
(2)本发明制备的羟基磷灰石/氧化石墨烯复合生物涂层显著提高了镁合金在模拟体液中的耐蚀性;并且羟基磷灰石与氧化石墨烯协同耦合,改善了镁合金基体中的电偶腐蚀,涂层腐蚀过程均匀可控,均匀分散在涂层中的氧化石墨烯具有抑菌作用;因此,这种复合涂层既可以提高镁合金基体的耐腐蚀性能,还能起到抗菌作用。
(3)本发明制备的羟基磷灰石/氧化石墨烯复合生物涂层厚度具有可控性,根据不同的电沉积时间,可以制备出不同厚度的涂层。
(4)本发明适用范围广。对于不同形状、尺寸的纯镁或镁合金试样,均可以在其表面制备羟基磷灰石/氧化石墨烯复合生物涂层。
附图说明
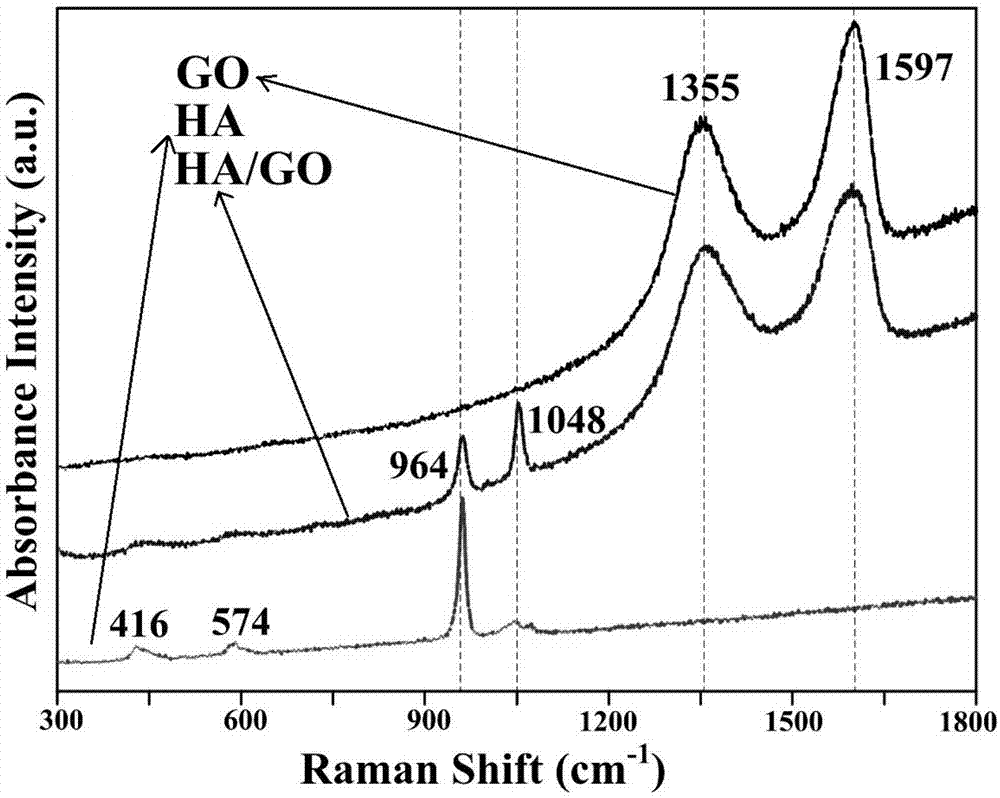
图1为本发明所述实施例1和对比例1得到的生物涂层以及单独的氧化石墨烯的拉曼图谱。
图2为本发明所述实施例1和对比例1得到的生物涂层以及单独的氧化石墨烯的傅里叶红外图谱。
图3为本发明所述实施例1得到的生物涂层的扫描电镜图片。
图4为本发明所述对比例1得到的生物涂层的扫描电镜图片。
图5为本发明所述对比例2得到的生物涂层的扫描电镜图片。
图6为本发明所述实施例1和对比例1得到的生物涂层以及没有沉积涂层的镁合金的动电位极化分析图。
图7为本发明所述实施例1和对比例1得到的生物涂层以及没有沉积涂层的镁合金的奈奎斯特图。
具体实施方式
下面结合附图对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
本发明提供一种医用镁合金表面羟基磷灰石和氧化石墨烯复合生物涂层的电沉积制备方法,包括如下步骤:
一种医用镁合金表面羟基磷灰石和氧化石墨烯复合生物涂层的电沉积制备方法,包括如下步骤:
步骤1:将纯镁或者镁合金进行预处理:
将纯镁或镁合金切割为20mm×15mm×5mm的长方体;
依次用400#、600#、1000#、1500#、2000#的水磨砂纸进行打磨;
丙酮超声清洗后,去离子水超声清洗5~10分钟,储存于无水乙醇中。
步骤2:将清洗后的纯镁或镁合金浸入3~6mol/lnaoh溶液中,浸泡30~120分钟,去离子水清洗后干燥。
步骤3:将羟基化的纯镁或镁合金在水解的氨丙基三乙氧基硅烷溶液中且室温下浸渍5~60分钟,然后在60~120℃下,加热30~60分钟,得到硅烷化的纯镁或者镁合金;
其中,所述氨丙基三乙氧基硅烷溶液含有的组分及其体积百分比为:硅烷:5%,乙醇:90%,去离子水:5%。
步骤4:配置氧化石墨烯电解液:
将0.02~0.05mol/l的ca(no3)2,0.01~0.03mol/l的nh4h2po4,2~6ml/l的h2o2,0.05~100μg/ml的氧化石墨烯加入去离子水中,配置成混合溶液;
将所述混合溶液经过超声处理,得到稳定存在的悬浮溶液。
步骤5:以硅烷化的纯镁或镁合金为阴极,石墨片为阳极,置于氧化石墨烯电解液中进行电沉积,在电沉积过程中,始终在磁力搅拌下进行沉积,得到具有羟基磷灰石和氧化石墨烯复合涂层的纯镁或者镁合金;
其中,所述氧化石墨烯电解液的ph为4~5,所述氧化石墨烯电解液的温度为55~65℃,所述电沉积的沉积电压为10~15v,沉积时为间0~60min。
步骤6:电沉积结束后,取出表面沉积有羟基磷灰石和氧化石墨烯复合生物涂层的纯镁或镁合金,自然晾干。
实施例1
在mg-1sn-1zn-0.5ca合金表面电沉积制备羟基磷灰石/氧化石墨烯复合生物涂层,制备步骤如下:
步骤1:基底材料准备:
将mg-1sn-1zn-0.5ca合金切割为20mm×15mm×5mm的长方体,用400#、600#、1000#、1500#、2000#的水磨砂纸依次进行打磨,采用丙酮超声清洗,去离子水超声清洗10分钟除去表面油污,之后储存于无水乙醇中待用。
步骤2:将清洗干净的mg-1sn-1zn-0.5ca合金浸入5mol/lnaoh溶液中,浸泡120分钟,然后用去离子水清洗、干燥。
步骤3:硅烷化处理:将5%(v/v)的硅烷,90%(v/v)的乙醇,5%(v/v)的水混合到一起,制备氨丙基三乙氧基硅烷(aptes)溶液,将该溶液在室温下搅拌60分钟,促进其进一步水解;将羟基化镁合金通过在水解的aptes溶液中室温下浸渍60分钟,然后在120℃下,加热30分钟。
步骤4:电沉积制备羟基磷灰石/氧化石墨烯复合涂层:a、配置电解液:0.042mol/lca(no3)2,0.025mol/lnh4h2po4,6ml/lh2o2,100μg/ml氧化石墨烯加入去离子水中,配置成混合溶液,将溶液经过超声处理,形成能够稳定存在的悬浮溶液,将电解液ph调为4.5,电解液温度为60℃;b、电沉积:以硅烷化mg-1sn-1zn-0.5ca合金为阴极,石墨片为阳极,沉积电压为10v,沉积时为间30min,在磁力搅拌条件下进行电沉积。电沉积结束后,把样品取出,自然晾干,得到羟基磷灰石/氧化石墨烯复合生物涂层(记为ha/go)该涂层的厚度为15μm。
实施例2
在az31镁合金表面电沉积制备羟基磷灰石/氧化石墨烯复合生物涂层,制备步骤如下:
步骤1:基底材料准备:
将az31合金切割为20mm×15mm×5mm的长方体,用400#、600#、1000#、1500#、2000#的水磨砂纸依次进行打磨,采用丙酮超声清洗,去离子水超声清洗10分钟除去表面油污,之后储存于无水乙醇中待用。
步骤2:将清洗干净的az31合金浸入3mol/lnaoh溶液中,浸泡120分钟,然后用去离子水清洗、干燥。
步骤3:硅烷化处理:将5%(v/v)的硅烷,90%(v/v)的乙醇,5%(v/v)的水混合到一起,制备氨丙基三乙氧基硅烷(aptes)溶液,将该溶液在室温下搅拌30分钟,促进其进一步水解;将羟基化镁合金通过在水解的aptes溶液中室温下浸渍5分钟,然后在60℃下,加热45分钟。
步骤4:电沉积制备羟基磷灰石/氧化石墨烯复合涂层:a、配置电解液:0.042mol/lca(no3)2,0.025mol/lnh4h2po4,2ml/lh2o2,100μg/ml氧化石墨烯加入去离子水中,配置成混合溶液,将溶液经过超声处理,形成能够稳定存在的悬浮溶液,将电解液ph调为4,电解液温度为55℃;b、电沉积:以硅烷化az31合金为阴极,石墨片为阳极,沉积电压为12v,沉积时为间5min,在磁力搅拌条件下进行电沉积。电沉积结束后,把样品取出,自然晾干,得到羟基磷灰石/氧化石墨烯复合生物涂层,该涂层的厚度为10μm。。
实施例3
在纯镁表面电沉积制备羟基磷灰石/氧化石墨烯复合生物涂层,制备步骤如下:
步骤1:基底材料准备:
将纯镁切割为20mm×15mm×5mm的长方体,用400#、600#、1000#、1500#、2000#的水磨砂纸依次进行打磨,采用丙酮超声清洗,去离子水超声清洗10分钟除去表面油污,之后储存于无水乙醇中待用。
步骤2:将清洗干净的纯镁浸入6mol/lnaoh溶液中,浸泡120分钟,然后用去离子水清洗、干燥。
步骤3:硅烷化处理:将5%(v/v)的硅烷,90%(v/v)的乙醇,5%(v/v)的水混合到一起,制备氨丙基三乙氧基硅烷(aptes)溶液,将该溶液在室温下搅拌120分钟,促进其进一步水解;将羟基化镁合金通过在水解的aptes溶液中室温下浸渍30分钟,然后在100℃下,加热60分钟。
步骤4:电沉积制备羟基磷灰石/氧化石墨烯复合涂层:a、配置电解液:0.042mol/lca(no3)2,0.025mol/lnh4h2po4,6ml/lh2o2,100μg/ml氧化石墨烯加入去离子水中,配置成混合溶液,将溶液经过超声处理,形成能够稳定存在的悬浮溶液,将电解液ph调为5,电解液温度为65℃;b、电沉积:以硅烷化纯镁为阴极,石墨片为阳极,沉积电压为15v,沉积时间为60min,在磁力搅拌条件下进行电沉积。电沉积结束后,把样品取出,自然晾干,得到羟基磷灰石/氧化石墨烯复合生物涂层,该涂层的厚度为14μm。
实施例4
在纯镁表面电沉积制备羟基磷灰石/氧化石墨烯复合生物涂层,制备步骤如下:
步骤1:基底材料准备:
将纯镁切割为20mm×15mm×5mm的长方体,用400#、600#、1000#、1500#、2000#的水磨砂纸依次进行打磨,采用丙酮超声清洗,去离子水超声清洗10分钟除去表面油污,之后储存于无水乙醇中待用。
步骤2:将清洗干净的纯镁浸入6mol/lnaoh溶液中,浸泡120分钟,然后用去离子水清洗、干燥。
步骤3:硅烷化处理:将5%(v/v)的硅烷,90%(v/v)的乙醇,5%(v/v)的水混合到一起,制备氨丙基三乙氧基硅烷(aptes)溶液,将该溶液在室温下搅拌120分钟,促进其进一步水解;将羟基化镁合金通过在水解的aptes溶液中室温下浸渍30分钟,然后在100℃下,加热60分钟。
步骤4:电沉积制备羟基磷灰石/氧化石墨烯复合涂层:a、配置电解液:0.042mol/lca(no3)2,0.025mol/lnh4h2po4,6ml/lh2o2,50μg/ml氧化石墨烯加入去离子水中,配置成混合溶液,将溶液经过超声处理,形成能够稳定存在的悬浮溶液,将电解液ph调为5,电解液温度为60℃;b、电沉积:以硅烷化纯镁为阴极,石墨片为阳极,沉积电压为15v,沉积时为间60min,在磁力搅拌条件下进行电沉积。电沉积结束后,把样品取出,自然晾干,得到羟基磷灰石/氧化石墨烯复合生物涂层,该涂层的厚度为11μm。
对比例1
在mg-1sn-1zn-0.5ca合金表面电沉积制备羟基磷灰石涂层,制备步骤如下:
步骤1:基底材料准备
将mg-1sn-1zn-0.5ca合金切割为20mm×15mm×5mm的长方体,用400#、600#、1000#、1500#、2000#的水磨砂纸依次进行打磨,采用丙酮超声清洗,去离子水超声清洗10分钟除去表面油污,之后储存于无水乙醇中待用。
步骤2:将清洗干净的mg-1sn-1zn-0.5ca合金浸入5mol/lnaoh溶液中,浸泡120分钟,然后用去离子水清洗、干燥。
步骤3:硅烷化处理:将5%(v/v)的硅烷,90%(v/v)的乙醇,5%(v/v)的水混合到一起,制备氨丙基三乙氧基硅烷(aptes)溶液,将该溶液在室温下搅拌60分钟,促进其进一步水解;将羟基化镁合金通过在水解的aptes溶液中室温下浸渍60分钟,然后在120℃下,加热30分钟。
步骤4:电沉积制备羟基磷灰石涂层:a、配置电解液:0.042mol/lca(no3)2,0.025mol/lnh4h2po4,6ml/lh2o2加入去离子水中,配置成混合溶液,将溶液经过超声处理,形成能够稳定存在的悬浮溶液,将电解液的ph调为4.5,电解液温度为60℃;b、电沉积:以硅烷化mg-1sn-1zn-0.5ca合金为阴极,石墨片为阳极,沉积电压为10v,沉积时间为30min,在磁力搅拌条件下进行电沉积。电沉积结束后,把样品取出,自然晾干,得到羟基磷灰石涂层(记为ha),该涂层的厚度为8μm。
对比例2
在mg-1sn-1zn-0.5ca合金表面电沉积制备羟基磷灰石/氧化石墨烯复合生物涂层,制备步骤如下:
步骤1:基底材料准备
将mg-1sn-1zn-0.5ca合金切割为20mm×15mm×5mm的长方体,用400#、600#、1000#、1500#、2000#的水磨砂纸依次进行打磨,采用丙酮超声清洗,去离子水超声清洗10分钟除去表面油污,之后储存于无水乙醇中待用。
步骤2:将清洗干净的mg-1sn-1zn-0.5ca合金浸入5mol/lnaoh溶液中,浸泡120分钟,然后用去离子水清洗、干燥。
步骤3:硅烷化处理:将5%(v/v)的硅烷,90%(v/v)的乙醇,5%(v/v)的水混合到一起,制备氨丙基三乙氧基硅烷(aptes)溶液,将该溶液在室温下搅拌60分钟,促进其进一步水解;将羟基化镁合金通过在水解的aptes溶液中室温下浸渍60分钟,然后在120℃下,加热30分钟。
步骤4:电沉积制备羟基磷灰石涂层:a、配置电解液:0.042mol/lca(no3)2,0.025mol/lnh4h2po4,6ml/lh2o2,120μg/ml氧化石墨烯加入去离子水中,配置成混合溶液,将溶液经过超声处理,形成能够稳定存在的悬浮溶液,将电解液的ph调为4.5,电解液温度为60℃;b、电沉积:以硅烷化mg-1sn-1zn-0.5ca合金为阴极,石墨片为阳极,沉积电压为10v,沉积时间为30min,在磁力搅拌条件下进行电沉积。电沉积结束后,把样品取出,自然晾干,得到羟基磷灰石/氧化石墨烯复合生物涂层,该涂层的厚度为12μm。
对实施例1(ha/go)和对比例1(ha)得到的生物涂层以及单独的氧化石墨烯(go)进行拉曼图谱分析,具体如图1所示,发现来自po43-基团的峰在416,574,964和1048cm-1的波数处观察到。在羟基磷灰石涂层及羟基磷灰石/氧化石墨烯复合生物涂层中都可以观察到po43-基团的峰(964和1048cm-1),而po43-基团的峰出现与ha(羟基磷灰石)相的存在有关,说明涂层中存在ha相。go(氧化石墨烯)相有两个明显的特征峰d峰和g峰,分别位于1355和1597cm-1处。在羟基磷灰石/氧化石墨烯复合生物涂层中,检测到go的特征峰,说明go被成功的沉积到镁合金表面。
对实施例1(ha/go)和对比例1(ha)得到的生物涂层以及单独的氧化石墨烯(go)进行傅里叶红外光谱分析,具体如图2所示,在3429cm-1处有强吸收峰,是由-oh的伸缩振动产生的,此吸收峰可能来自于羟基磷灰石、氧化石墨烯吸附的水分子。568和601cm-1为po43-基团的o-p-o吸收峰,1099和1037为po43-基团的p-o吸收峰。1722cm-1左右吸收峰是由羧基的c-o和c=o的伸缩振动产生。1622cm-1左右的吸收峰属于石墨的c=c键或残余水分子的振动。可以看出在羟基磷灰石/氧化石墨烯复合生物涂层中存在由c=c和c-o的伸缩振动产生的吸收峰,而羟基磷灰石涂层中没有这些特征峰,证明go已经成功的负载在涂层中。
对实施例1和对比例1得到的生物涂层进行扫描电镜分析,具体如图3和图4所示,对比发现,单纯的羟基磷灰石涂层,其结构为粗大的片状结构,且表面起伏不平,均匀性较差;通过在镁合金表面共同沉积羟基磷灰石和氧化石墨烯,制备出的羟基磷灰石/氧化石墨烯复合生物涂层,微观结构明显细化,且更加均匀致密,即加入氧化石墨烯后,氧化石墨烯为羟基磷灰石提供了更多的形核位置,使羟基磷灰石的结构由粗大的片状结构变为细小的纳米针叶状结构,且氧化石墨烯均匀的分散在涂层中,整体涂层更加均匀致密,有效的提高了涂层与基体的结合强度;这种细小的纳米针叶状结构,可以促进细胞的黏附,有效的改善了镁合金基体的生物相容性。还为其优异的耐腐蚀性,提供了有效的保障。
通过实施例1和对比例2得到的生物涂层进行扫描电镜分析发现,如图3和图5所示,当氧化石墨烯的浓度超过120μg/ml时,制备的羟基磷灰石/氧化石墨烯复合生物涂层,微观结构并没有明显细化,也不够致密均匀。说明氧化石墨烯的量并不是越大越好,需要控制在一个合理的范围内。
对实施例1和对比例1得到的生物涂层以及没有沉积涂层的镁合金进行动电位极化分析,具体如图6所示,可以看出相比于镁合金基体,经过电沉积后的涂层试样的腐蚀电流密度(icorr)均有明显降低,而共同沉积羟基磷灰石和氧化石墨烯后,其腐蚀电流密度进一步降低。说明加入go可改善镁合金基体在模拟体液中的耐蚀性。
对实施例1和对比例1得到的生物涂层以及没有沉积涂层的镁合金进行奈奎斯特图分析,具体如图7所示,可以看出,经过电沉积制备涂层之后,其阻抗弧半径明显增大,说明耐腐蚀性有了明显的增加,而共同沉积羟基磷灰石和氧化石墨烯后,其阻抗弧半径增大更多,说明电沉积制备的羟基磷灰石/氧化石墨烯复合生物涂层具有最好的耐腐蚀性能。
本发明制备的羟基磷灰石/氧化石墨烯复合生物涂层显著提高了镁合金在模拟体液中的耐蚀性;并且羟基磷灰石与氧化石墨烯协同耦合,改善了镁合金基体中的电偶腐蚀,涂层腐蚀过程均匀可控,均匀分散在涂层中的氧化石墨烯具有抑菌作用;因此,这种复合涂层既可以提高镁合金基体的耐腐蚀性能,还能起到抗菌作用。
本发明克服了单一涂层的不足,提供了一种在纯镁或镁合金表面电沉积制备复合生物涂层的方法,并且该制备方法简单、效率高、易操作、适合大面积生产推广。制备出的羟基磷灰石/氧化石墨烯复合生物涂层,微观结构明显细化,且更加均匀致密。这为其优异的耐腐蚀性,提供了有效的保障;而且在加入氧化石墨烯后,有效的提高了涂层与基体的结合强度。还能根据不同的电沉积时间,可以制备出不同厚度的涂层。
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
- 还没有人留言评论。精彩留言会获得点赞!