一种绿色电化学偶联制备茶碱类衍生物的方法
14296)采用pt丝做阳极,pt片做阴极,单池条件下实现了苯胺与γ-内酰胺的c-h/n-h接氧化脱氢偶联。通过循环伏安研究发现对甲基苯胺的氧化电势(0.77v vs ag/ag
+
)明显低于n-甲基吡咯烷酮(nmp)的氧化电势(1.90v vs ag/ag
+
),而此反应仍然能在同一个体系中发生二者的同时氧化。作者通过控制实验推测可能是因为阳极采用铂丝后氧化端接触面积减少,电流密度显著增强,此时溶剂计量的nmp浓度远远高于反应底物4-甲基苯胺,从而增大了nmp被氧化的机会。尽管n-脱甲基反应的方式多种多样,但均不能完全克服底物适用性不广泛,使用昂贵、有毒、难合成配合物型试剂,反应条件苛刻(无水、无氧等)或者操作不够便利等缺陷。近年来,电化学有机合成以清洁电子做氧化还原试剂参加化学反应,实现化学的反应的低污染甚至零污染,在绿色化学领域优势逐渐凸显。2017年,lei等(wu,j.zhou,y.chiang,c.w.lei,a.electro-oxidative c(sp3)-h amination of azoles via intermolecular oxidative c(sp3)-h/n-h cross-coupling.acs catal.,2017,7(12),8320-8323)通过阳极直接氧化csp3-h与唑类n-h脱氢交叉偶联。该组在乙腈溶液中加入电解质四丁基氟硼酸铵(tbabf4),并在80℃下以12ma恒流电解得到产物。初步研究了反应机理,推测可能为自由基反应类型。此反应适用性较好,苯并三氮唑与thf反应产率高达92%,苯环上取代单甲基、二甲基产率也较为理想;取代-coome时,产率下降至60%。其它三氮唑、四氮杂环、噁唑产率也多在80%-90%。当选择苯并三氮唑做n源时,发现nmp的α-位c-h与之脱氢偶联的产物产率也较为理想,为85%。其它杂环醚则产率在42%-63%之间。
4.2018年,ackermann等(qiu,y.struwe,j.meyer,t.h.oliveira,j.c.a.ackermann,l.catalyst-and reagent-free electrochemical azole ch amination chem.eur.j.,2018,24(49),12784-12789)用阳极氧化吗啉n-h和苯并恶唑n-h失去h
+
,继而交叉偶联,同时阴极还原释放h2。此反应为室温下,简单、高效、便捷的单池、恒流电解反应。通过底物拓展,n-甲基苯胺、二烯丙基胺、n-甲基-n-苄基胺、二乙基胺、环状二级胺等都能获得不错的产率,在50%-95%之间。
5.zeng等(lian,f.sun,c.xu,k.zeng,c.electrochemical dehydrogenative imidation of n-methyl substituted benzylamines with phthalimides for the direct synthesis of phthalimide-protected gem-diamines.org.lett.,2019,21(1),156-159)直接电氧化csp
3-h使n,n-二甲基苄胺氧化形成c=n双键,同时meoh溶剂在阴极还原产生的meo-夺取邻苯二甲酰亚胺氮上的氢,使之对c=n双键进行亲核加成反应。除了邻苯二甲酰亚胺,苯并三氮唑、三氮唑均有较好的反应活性,产物产率分别为83%,79%。此反应除了氢气释放,也无其它副产物生成。2018年,lei等(tang,s.wang,s.liu,y.cong,h.lei,a.electrochemical oxidative ch amination of phenols:access to triarylamine derivatives.angew.chem.int.ed.,2018,57(17),4737-4741)也对苯酚的电化学进行了研究,他们发现使用ni做阴极,可以实现苯酚的芳环c-h与n-h交叉脱氢偶联。反应用时较短,均在100分钟左右即可以完成,而且产率较高。通过底物拓展可以看出大部分产物产率集中在70%-90%。容易氧化的酚羟基在电化学氧化体系中可以很容易保留下来,这说明电化学条件可以比较温和,也可以做到选择性氧化。黄精美课题组(a.du k s,huang j m,synthesis of bisindolylmethanes from indoles and ethers[j],org.lett.,2018,20,2911-2915;b.黄精美,杜克斯,1,1
’‑
二吲哚甲烷类衍生物的电化学合成方法[p],中国专利号:zl201610930550.2;c.杜克斯,电化学x-h的直接氧化偶联以及偶
氮化合物的合成[d],华南理工大学博士论文,2019.)研究电化学条件下醚的双吲哚化反应。该方法通过催化量的lewis酸lacl3作为媒介,liclo4作为导电盐,在室温、大气环境下以5ma的恒电流在相应的溶剂中进行电解反应,实现了各种取代的吲哚衍生物对醚的双吲哚氧化偶联。该方法反应条件温和,操作简单。通过对吲哚环5-位引入吸电子基和给电子基进行底物适用性研究发现其官能团兼容性较好,均取得较高的产率。
技术实现要素:
[0006]
本发明的目的在于提供一种绿色电化学偶联制备茶碱类衍生物的方法,解决现有技术中茶碱类衍生物制备方案废弃物排放多,环境污染严重等技术问题。
[0007]
本发明公开了一种绿色电化学偶联制备茶碱类衍生物的方法,包括以下步骤:
[0008]
s1.将茶碱(或咖啡因)溶于溶剂中;
[0009]
s2.加入催化剂和电解质,插入电极,调节电极大小和极间距离,室温下搅拌并通电产生反应;
[0010]
s3.反应完全后,使用柱层析法分离纯化;
[0011]
s4.将纯化后的物质重结晶后得到产物。
[0012]
进一步的,所述茶碱的浓度为0.1—1.0mmol/ml。
[0013]
进一步的,所述茶碱的结构式为:
[0014][0015]
进一步的,所述溶剂为乙腈和饱和thf(四氢呋喃)溶液的混合溶剂。
[0016]
进一步的,所述混合溶剂中乙腈与thf的体积比为1:3-3:1,同时thf也作为原料参与反应。
[0017]
进一步的,所述催化剂为六水合硝酸镧。
[0018]
进一步的,所述催化剂的量为茶碱物质的量的5%—20%。
[0019]
进一步的,所述电解质为高氯酸锂。
[0020]
进一步的,所述高氯酸锂的浓度为0.02-2.0mol/ml。
[0021]
进一步的,所述电极包括阳极和阴极,阴极和阳极距离可选为10~15mm。
[0022]
进一步的,所述步骤s2中反应电流强度为1~10ma。
[0023]
进一步的,所述步骤s2中反应电流强度为2~5ma。
[0024]
进一步的,所述步骤s2中通电反应时间为1-10h。
[0025]
进一步的,所述步骤s4中重结晶使用乙腈作为溶剂。
[0026]
一种绿色电化学偶联制备茶碱类衍生物的方法制备的茶碱衍生物,其结构式为:
[0027][0028]
本发明的有益效果为:
[0029]
1.本发明通过电化学手段催化反应,避免了使用化学计量的传统氧化剂,从而避免各种废弃物的排放,减少环境污染,成本低廉;
[0030]
2.本发明条件温和,室温即可发生反应整个操作过程仅需要在传统的搅拌反应装置上通上直流电,简单易行,成本低,污染小,符合绿色化学的理念。
附图说明
[0031]
为了更清楚地说明本发明实施方式的技术方案,下面将对实施方式中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施方式,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
[0032]
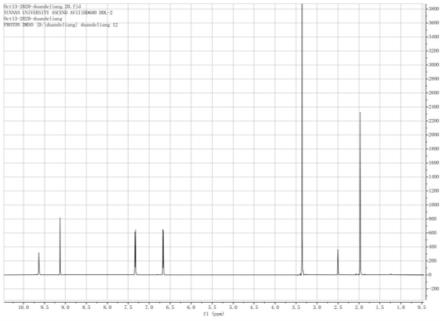
图1为本发明实施案例1制备的产物1h-nmr图谱;
[0033]
图2为本发明实施案例1制备的产物
13
c-nmr图谱;
[0034]
图3为本发明实施案例1制备的产物esi-ms正离子条件下质谱图;
[0035]
图4为本发明实施案例1制备的产物esi-ms负离子条件下质谱图;
[0036]
图5为本发明实施案例2制备的产物hplc-esi-ms正离子条件下质谱图;
[0037]
图6目标化合物结构示意图。
具体实施方式
[0038]
需要说明的是,在不冲突的情况下,本发明中的实施方式及实施方式中的特征可以相互组合。
[0039]
实施例1
[0040]
本发明的实施方式提供了一种绿色电化学偶联制备茶碱类衍生物的方法,具体步骤如下:
[0041]
向10ml试管中依次加入144.2mg(0.8mmol)茶碱,34.64mg(0.08mmol)六水合硝酸镧,106.4mg(0.2mol/ml)liclo4;然后加入四氢呋喃4ml,乙腈1ml,插入铂片电极和铂丝电极,电极距离为10mm,阴极为铂片电极,阳极为铂丝电极,直流电源供电5ma,6.5h反应完全。反应液用硅胶柱层析进行分离,洗脱剂为环己烷:乙酸乙酯=1:1,随着洗脱进程,逐渐增大乙酸乙酯的比例,增大极性,加快洗脱进程。将分离液分别旋转蒸发,冷却结晶,使用乙腈进行重结晶。反应原理如下:
[0042][0043]
以上实施例得到的产物1h nmr图(图1)、
13
cnmr图(图2)、正离子条件下esi-ms图(图3)、负离子条件下esi-ms图(图4)如附图所示,鉴定数据如下:
[0044]1h nmr(600mhz,dmso):δ9.63,9.12,7.33,6.65,3.33,1.97ppm。
[0045]
13
cnmr(600mhz,dmso):δ167.8,153.45,131.3,120.78,115.7,39.52(dmso),23.8ppm。
[0046]
图1、图2、图3、图4结合分析实施例得到的产物结构与目标产物一致,如下所示:
[0047][0048]
该实施例制备得到的新的茶碱衍生物的1h nmr图(图1),1h nmr(600mhz,dmso):δ9.63,9.12,7.33,6.65,3.33,1.97ppm.表明具有茶碱的特征结构。该实施例制备得到的新的茶碱衍生物的
13
cnmr图(图2)
13
cnmr(600mhz,dmso):δ167.8,153.45,131.3,120.78,115.7,39.52(dmso),23.8ppm表明具有茶碱的特征结构。
[0049]
该实施例制备得到的新的茶碱衍生物的质谱esi-ms正离子峰图谱如图3所示,其中m/z 452为样品的m+na峰,m/z 436为样品的m+4h峰,m/z 397为样品的碎片的茶碱+3个四氢呋喃的峰,m/z 325为样品的的碎片的茶碱+2个四氢呋喃的峰,m/z 309为样品中的碎片的茶碱+2个四氢呋喃去掉一个氧的峰。
[0050]
该实施例制备得到的新的茶碱衍生物的质谱esi-ms负离子峰图谱如图4所示esi-ms负离子峰中m/z 196为样品的的碎片的茶碱+1个氧的峰。
[0051]
结合以上分析实施例得到的产物结构与目标产物一致。
[0052]
实施例2
[0053]
本发明的实施方式提供了一种绿色电化学偶联制备咖啡因衍生物2的方法的制备方法,具体步骤如下:
[0054]
向10ml试管中依次加入155.4mg(0.8mmol)咖啡因,34.64mg(0.08mmol)六水合硝酸镧,106.4mg(0.2mol/ml)liclo4;然后加入四氢呋喃4ml,乙腈1ml,控制温度为100℃,插入铂片电极和铂丝电极,电极距离为10mm,阴极为铂片电极,阳极为铂丝电极,直流电源供电5ma,6.5h反应完全。反应液用硅胶柱层析进行分离,洗脱剂为环己烷:乙酸乙酯=1:1,随着洗脱进程,逐渐增大乙酸乙酯的比例,增大极性,加快洗脱进程。将分离液分别旋转蒸发,冷却结晶,使用乙腈进行重结晶。反应原理如下:
[0055][0056]
以上实施例得到的产物hplc-ms正离子条件下esi-ms图(图5)如附图所示,鉴定数据如下:该实施例制备得到的新的咖啡因衍生物2的质谱esi-ms正离子峰图谱如图5所示,其中m/z 452为样品的m+h峰m/z 461.15。
[0057]
同时得到新的偶联化合物3和4,hplc-ms质谱波谱esi正离子峰m/z 377.16、355.19解析为两种新的双偶联产物3、4的分子离子峰。
[0058]
本发明不局限于上述可选实施方式,任何人在本发明的启示下都可得出其他各种
形式的产品,但不论在其形状或结构上作任何变化,凡是落入本发明权利要求界定范围内的技术方案,均落在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1