一种原位生长在N-C骨架内的Co4N量子点电极材料的制备方法
一种原位生长在n-c骨架内的co4n量子点电极材料的制备方法
技术领域
1.本发明属于电催化材料领域,涉及一种原位生长在n-c内的co4n量子点电极材料的制备方法,具体地说,是涉及一种用于全ph值电解水制氢、制氧的嵌于n掺杂碳骨架中的co4n量子点原位生长在碳布上的自支撑复合电极的制备方法。
背景技术:
2.近年来,由于氢气新能源产业的不断发展,电解水制氢技术的开发应用越来越受到科研人员的重视。如何制备一种性能高效、价格低廉、能在宽ph值范围应用的电催化材料成为氢能生产应用的关键,在诸多电催化材料中,贵金属pt基催化剂是水分解析氢反应(her)最高效的电催化剂,而iro2和ruo2对水分解析氧反应(oer)展现出了最优的催化效率,但是,有限的资源及过高的成本制约了这些贵金属催化剂在市场上的大量应用。因此,合成并且开发新型高效的非贵金属多过渡金属化合物催化剂,以取代资源稀少价格高的贵金属催化剂,对于电解水制备清洁的氢气新能源应用有着重要的研究意义。然而这些催化剂也和贵金属催化剂一样,常常只能进行单一的催化反应,有的在析氢反应中表现出优异的性能而在析氧反应中性能一般,难以在同一种电解质溶液中同时在析氢反应和析氧反应中表现出优异的性能。这就会导致催化剂的制作成本提高。因此,迫切需要开发可在宽ph范围内通用的双功能电催化剂以适应多样化的应用。
3.另外,值得注意的是,大多数oer催化剂要么在强酸性溶液中容易遭受腐蚀,要么在酸性介质中表现出较差的活性。另外,中性电解质对环境无害且具有生物相容性,不幸的是,很少有催化剂在中性介质中表现良好。因此,迫切需要开发可在宽ph范围内通用的双功能电催化剂以适应多样化的应用。
4.目前,只有少数非贵金属催化剂用于宽ph全解水,制备方法要么繁琐,要么稳定性差。过渡金属氮化物co4n已用于在碱性电解质中进行her反应,然而,其制备方法要么繁琐,要么需要毒性大,易泄露氨气作n源,因此,迫切需要开发一种简单,环保,经济的制备方法来制备高效、稳定的在宽ph范围内使用的高效双功能电催化剂,用于大规模工业化使用。
5.同时,得到的氮化物电催化材料粉体需要用胶黏剂粘附在集流体的表面,催化剂/胶黏剂/集流体两个界面的存在会降低电极的导电性和稳定性。因此,设计合成具有优良催化性能的自支撑氮化钴电催化材料,用于电解水制氢反应,具有重要的实用价值和现实意义。
技术实现要素:
6.本发明针对现有技术中,宽ph全解水所需电极材料制备方法繁琐,需要使用毒性大,易泄露氨气作n源进行氮化,成本高,稳定性差等缺点,提出了一种用于全ph值(酸性、中性和碱性)电解水制氢、制氧的原位生长在n-c骨架内的co4n量子点电极材料的制备方法,其特征在于,通过液体前驱体对碳布的高度渗透,使植入生长、碳化和co4n量子点原位高度分散一步完成,得到原位生长在n-c骨架中的co4n量子点电极材料,将其用于宽ph全解水制
氢,制氢效率高,所述电极材料的制备方法包括下述步骤:
7.本发明针对现有技术中电催化剂难以在广泛ph值范围都表现处高的制氢活性,co4n复合电催化材料的制备过程复杂、成本高,电极的稳定性差等缺点,提出了一种嵌于n掺杂碳骨架中的co4n量子点/碳布自支撑复合电极的制备方法。所述电极是一种电催化材料原位生长于碳布集流体上的自支撑型复合电极,所述电催化材料是原位生长于碳布集流体上的嵌于n掺杂碳骨架中的co4n量子点。
8.(1)先将碳布裁剪为1
×
1-20
×
20cm2,再将裁剪好的碳布依次用丙酮、5%hcl溶液、去离子水、乙醇各超声处理15min,然后将碳布浸入h2so4/h2o2混合溶液中12-48h并用去离子水洗涤,将预处理的碳布在氮气流下干燥;
9.(2)将0.01-1mol固体cocl2·
6h2o和0.01-1mol固体丙二酸混合均匀,在40-80℃油浴中搅拌加热形成均匀透明深度共熔溶液;
10.(3)取0.01-10ml溶液均匀涂布于步骤(1)得到的碳布表面,置于石英舟中,在通入n2气流的管式炉中加热,并在n2气流上游区另一石英舟中加入1.0-100.0g尿素,在n2气氛下以1-10℃
·
min-1
升温至700-900℃,并保温1-6h,得到所述电极材料。
11.本发明的优点在于:该方法工艺简单,通过液体前驱体对碳布的高度渗透,使植入生长碳化和co4n量子点原位高度分散一步完成,得到原位生长在n-c内的co4n量子点电极材料,co在界面处与c形成了新的化学键,这种化学键在界面处有利于电子的快速转移,从而提高电导率并提高催化活性。基底材料的引入可以实现催化剂的自支撑,避免了粘接剂的使用,从而避免了非活性位点的产生,并且基底材料直接作为电极,催化剂与基底材料的直接接触将会促进电子的转移从而进一步改善其导电性。原位生长在n-c内的co4n量子点电极材料能保证电子从基底到表面的高效传输,使制备的电极材料在宽ph具有优异的her、oer和全解水性能和稳定性。对有机精细化学品也有很好的电催化氧化选择性。对电催化氧化甲醛也有很好的催化活性。通过电催化氧化用于工业排放的有害气体h2s,co,no,nh3及汽车尾气的绿色净化。
附图说明
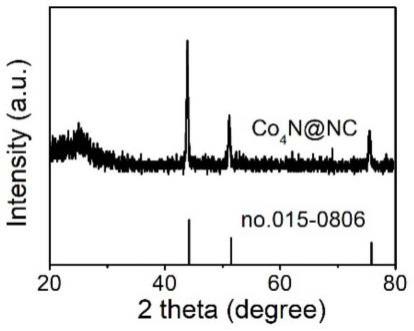
12.图1为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料的xrd谱图;
13.图2为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料的不同放大倍数的sem照片;
14.图3为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料相应的元素sem-mapping图谱;
15.图4为利用本发明实施例一所述方法经步骤(2)制备的原位生长在n掺杂c骨架中的co4n量子点电极材料不同放大倍数的hrtem照片;
16.图5为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料的拉曼谱图;
17.图6为利用本发明对比例一所述方法制备的co4n电极材料的不同放大倍数的sem照片;
18.图7为利用本发明对比例四所述方法制备的co4n@c电极材料的sem照片;
19.图8为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料(a)和本发明对比例一所述方法制备的co4n电极材料(b)对气泡的接触角实验结果;
20.图9在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一碳布对比例四的her极化曲线;
21.图10在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料在相应her电压下的稳定性测试结果;
22.图11在1.0m氢氧化钾中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料和对比例一电极材料的her极化曲线;
23.图12在1.0m pbs中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料和对比例一电极材料的her极化曲线;
24.图13在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一电极材料和对比例二iro2的oer极化曲线;
25.图14在1.0m氢氧化钾中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一电极材料和对比例二iro2的oer极化曲线;
26.图15在1.0m pbs中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一电极材料和对比例二iro2的oer极化曲线;
27.图16在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料在相应oer电压下的稳定性测试;
28.图17在1.0m氢氧化钾中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料在相应oer电压下的稳定性测试;
29.图18在1.0m pbs中,本发明实施例一所述方法制备的原位生长在n-c框架内的co4n量子点电极材料在相应oer电压下的稳定性测试;
30.图19在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料作为her和oer双功能催化剂用于全解水和对比例三pt/c、iro2双电极全解水的lsv曲线;
31.图20在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料作为her和oer双功能催化剂用于全解水的长期稳定性测试。
32.图21在1.0m koh中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料作为her和oer双功能催化剂用于全解水和对比例三pt/c、iro2双电极全解水的lsv曲线。
33.图22在1.0m pbs中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料作为her和oer双功能催化剂用于全解水和对比例三pt/c、iro2双电极全解水的lsv曲线。
具体实施方式
34.下面通过实施例和对比例对本发明作进一步详细说明:
35.实施例一:
36.(1)先将碳布裁剪为1
×
1cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离
子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中48h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
37.(2)将0.01mol cocl2·
6h2o和0.03mol丙二酸混合均匀,在60℃油浴中搅拌加热形成均匀透明溶液;
38.(3)取0.2ml溶液均匀涂布于1
×
1cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉加热上游区另一石英舟中加入8.0g尿素,在n2气氛下以5℃
·
min-1
升温至850℃保温4h。
39.实施例二:
40.(1)先将碳布裁剪为1
×
1cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中48h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
41.(2)将0.01mol cocl2·
6h2o和0.03mol丙二酸混合均匀,在70℃油浴中搅拌加热形成均匀透明溶液;
42.(3)取0.2ml溶液均匀涂布于1
×
1cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉加热上游区另一石英舟中加入4.0g尿素,在n2气氛下以5℃
·
min-1
升温至850℃保温4h。
43.实施例三:
44.(1)先将碳布裁剪为1
×
1cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中48h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
45.(2)将0.01mol cocl2·
6h2o和0.06mol丙二酸混合均匀,在60℃油浴中搅拌加热形成均匀透明溶液;
46.(3)取0.05ml溶液均匀涂布于1
×
1cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉加热上游区另一石英舟中加入2.0g尿素,在n2气氛下以10℃
·
min-1
升温至850℃保温4h。
47.实施例四:
48.(1)先将碳布裁剪为1
×
1cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中48h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
49.(2)将0.01mol cocl2·
6h2o和0.03mol丙二酸混合均匀,在60℃油浴中搅拌加热形成均匀透明溶液;
50.(3)取0.2ml溶液均匀涂布于1
×
1cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉加热上游区另一石英舟中加入6.0g尿素,在n2气氛下以5℃
·
min-1
升温至800℃保温6h。
51.实施例五:
52.(1)先将碳布裁剪为6
×
6cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中24h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
53.(2)将0.03mol cocl2·
6h2o和0.03mol丙二酸混合均匀,在60℃油浴中搅拌加热形
成均匀透明溶液;
54.(3)取1ml溶液均匀涂布于6
×
6cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉加热上游区另一石英舟中加入10.0g尿素,在n2气氛下以5℃
·
min-1
升温至850℃保温4h。
55.实施例六:
56.(1)先将碳布裁剪为8
×
8cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中48h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
57.(2)将0.02mol cocl2·
6h2o和0.02mol丙二酸混合均匀,在80℃油浴中搅拌加热形成均匀透明溶液;
58.(3)取2ml溶液均匀涂布于8
×
8cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉加热上游区另一石英舟中加入8.0g尿素,在n2气氛下以2℃
·
min-1
升温至850℃保温4h。
59.实施例七:
60.(1)先将碳布裁剪为15
×
15cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中12h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
61.(2)将0.5mol cocl2·
6h2o和1mol丙二酸混合均匀,在60℃油浴中搅拌加热形成均匀透明溶液;
62.(3)取3ml溶液均匀涂布于15
×
15cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉加热上游区另一石英舟中加入25.0g尿素,在n2气氛下以5℃
·
min-1
升温至900℃保温1h。
63.对比例一:
64.(1)先将碳布裁剪为1cm2,再将裁剪好的碳布依次用丙酮、hcl溶液v、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中48h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
65.(2)将0.01mol cocl2·
6h2o溶于水,均匀涂布于1cm2步骤(1)得到的碳布表面,干燥,置于石英舟中,在nh3气氛下以5℃
·
min-1
升温至850℃保温4h。
66.对比例二:
67.(1)将20mg iro2分散在0.5ml去离子水、0.45ml乙醇和50μl nafion的混合溶液中,在超声辅助下分散均匀;
68.(2)量取180μl步骤(1)所得混合液滴涂在1
×
1cm2的碳布基底上,室温干燥,得到iro2电极。
69.对比例三:
70.(1)称取20mg pt/c,加入0.5ml去离子水和0.45ml乙醇超声混合均匀,然后,混合液中加入50μl nafion继续超声分散30min,得到1ml混合均一的溶液,量取180μl的混合溶液均匀滴涂在1
×
1cm2的碳布基底上,将其干燥得到pt/c电极;
71.(2)称取20mg iro2,加入0.5ml去离子水和0.45ml乙醇超声混合均匀,然后,混合液中加入50μl nafion继续超声分散30min,得到1ml混合均一的溶液,量取180μl的混合溶
液均匀滴涂在1
×
1cm2的碳布基底上,将其干燥得到iro2电极;
72.(3)将步骤(1)得到的pt/c电极作阴极,将步骤(2)得到的iro2电极作阳极,用于全解水分解。
73.对比例四:
74.(1)先将碳布裁剪为1
×
1cm2,再将裁剪好的碳布依次用丙酮、hcl溶液(5%)、去离子水、乙醇各超声15min,然后将碳布浸入h2so4/h2o2溶液中48h并用超纯水洗涤,然后将预处理的碳布在氮气流下干燥;
75.(2)将0.01mol cocl2·
6h2o和0.03mol丙二酸混合均匀,在60℃油浴中搅拌加热形成均匀溶液;
76.(3)取0.2ml溶液均匀涂布于1
×
1cm2步骤(1)得到的碳布表面,置于石英舟中,在管式炉在nh3气氛下以5℃
·
min-1
升温至850℃保温4h。
77.图1为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料的xrd谱图。与金属co的标准卡相比,co4n@nc的所有特征衍射峰都向较低的衍射角偏移。这可能是由于n原子掺入到co晶格中,导致了晶格的膨胀。
78.图2为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料的不同放大倍数的sem照片。从图可以看出负载催化剂后,碳布的表面变得粗糙(图2a)。进一步放大的sem图像(图2b,图2c)显示负载在碳布表面上的催化剂由小纳米颗粒组成。与此形成鲜明对比的是,对比例一直接通过氨气与cocl2·
6h2o氨化得到的co4n具有非常不均匀的形貌和块状结构(图6),这充分表明本发明所采用的制备方法可以有效且可控地合成形貌均匀的纳米材料。
79.图3为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料相应的元素sem-mapping图谱。相应的元素的stem-mapping证实了co、n和c元素的均匀分布。
80.图4为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料的tem照片(a)和hrtem照片(b)。负载在碳布表面的催化剂由超小量子点组成,其尺寸非常均匀,平均粒径约为3nm。小而均匀的量子点有利于暴露出更多的不饱和位点,极大地有助于催化剂催化活性的提高。hrtem照片中标识的d间距为0.205nm的晶格条纹,与co4n的(111)面一致。
81.图5为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料的拉曼谱图。显示出无定型碳的两个特征峰。
82.图6为利用本发明对比例一所述方法制备的co4n电极材料的不同放大倍数的sem照片;从图可以看出,所得co4n形貌不规整,大小不一,约1-10μm。
83.图7为利用本发明对比例四所述方法制备的co4n@c电极材料的sem照片;从图可以看出,所得co4n@c形貌成不规则块状,大小不一,约100-200nm。
84.图8为利用本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料(a)和本发明对比例一所述方法制备的co4n电极材料(b)对气泡的接触角实验结果。从图可以看出,本发明实施例一所述方法制备电极材料对气泡的接触角为140.6℃,远大于对比例一电极材料对气泡的接触角,说明本发明实施例一所述方法制备电极材料疏气性好于对比例一电极材料。
85.图9是在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一和对比例四电极材料的her极化曲线;可以看出,在酸性电解质中,实施例一电极材料在56mv的低过电位下即可达到10ma cm-2
的电流密度,远优于对比例一、对比例四电极材料的her性能。
86.图10是在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料在相应her电压下的稳定性测试,高电流下,电流密度长时间保持不变,表现出很好的稳定性。
87.图11是在1.0m氢氧化钾中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料和对比例一电极材料的her极化曲线。在碱性电解质中,实施例一电极材料在72mv的低过电位下达到10ma cm-2
的电流密度,远低于对比例一电极材料her的过电位。
88.图12是在1.0m pbs中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料和对比例一电极材料的her极化曲线,在中性电解质中,实施例一电极材料在174mv的低过电位下达到10ma cm-2
的电流密度,远低于对比例一电极材料her的过电位。
89.图13是在0.5m h2so4中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一电极材料和对比例二iro2的oer极化曲线;在酸性电解质中,实施例一电极材料所得oer在110mv的低过电位下达到10ma cm-2
的电流密度,性能优于对比例一电极材料和对比例二iro2电极材料。
90.图14是在1.0m氢氧化钾中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一电极材料和对比例二iro2的oer极化曲线。在碱性电解质中,实施例一所得oer在120mv的低过电位下分别达到10ma cm-2
的电流密度,性能优于对比例一电极材料和对比例二iro2电极材料。
91.图15是在1.0m pbs中本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料、对比例一电极材料和对比例二iro2的oer极化曲线。在中性电解质中,实施例一所得oer在195mv的低过电位下分别达到10ma cm-2
的电流密度,性能优于对比例一电极材料和对比例二iro2电极材料。
92.图16是在0.5m h2so4中本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料在相应oer电压下的稳定性测试,表现出很好的稳定性。
93.图17是在1.0m氢氧化钾中本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料在相应oer电压下的稳定性测试,表现出很好的稳定性。
94.图18是在1.0m pbs中本发明实施例一所述方法制备的原位生长在n-c框架内的co4n量子点电极材料在相应oer电压下的稳定性测试,表现出很好的稳定性。
95.图19是在0.5m h2so4中本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料作为her和oer双功能催化剂用于全解水和对比例三pt/c、iro2双电极全解水的lsv曲线。原位生长在n掺杂c骨架中的co4n量子点电极材料表现出出色的整体水分解能力,在酸性电解质中仅需要1.47v较低的电位即可获得10ma cm-2
的电流密度,性能优于对比例三pt/c、iro2双电极全解水性能。
96.图20是在0.5m h2so4中本发明实施例一所述方法制备的原位生长在n掺杂c骨架中
的co4n量子点电极材料作为her和oer双功能催化剂用于全解水的长期稳定性测试,表现出很好的稳定性。
97.图21是1.0m koh中本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料作为her和oer双功能催化剂用于全解水和对比例三pt/c、iro2双电极全解水的lsv曲线。原位生长在n掺杂c骨架中的co4n量子点电极材料表现出出色的整体水分解能力,在碱性电解质中仅需要1.48v较低的电位即可获得10ma cm-2
的电流密度,性能优于对比例三pt/c、iro2双电极全解水性能。
98.图22是在1.0m pbs中,本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料作为her和oer双功能催化剂用于全解水和对比例三pt/c、iro2双电极全解水的lsv曲线。原位生长在n掺杂c骨架中的co4n量子点电极材料表现出出色的整体水分解能力,在中性电解质中仅需要1.61v较低的电位即可获得10ma cm-2
的电流密度,性能优于对比例三pt/c、iro2双电极全解水性能。
99.本发明实施例一所述方法制备的原位生长在n掺杂c骨架中的co4n量子点电极材料,对有机精细化学品也有很好的电催化氧化选择性。对电催化氧化甲醛也有很好的电化学催化活性。通过电催化氧化用于工业排放的有害气体h2s,co,no,nh3及汽车尾气的绿色净化。
100.上述实施例是本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,未背离本发明的原理与工艺过程下所作的其它任何改变、替代、简化等,均为等效的置换,都应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1