一种利用微通道反应装置连续制备烷基化异喹啉酮类化合物的方法
1.本发明属于有机合成领域,具体涉及一种利用微通道反应装置连续制备烷基化异喹啉酮类化合物的方法。
背景技术:
2.异喹啉二酮及其衍生物是一类非常重要的含氮杂环化合物,广泛存在于天然产物、药物分子及有机功能材料中,研究表明这类化合物具有明显的生理活性,如具有抗肿瘤、抗镇痛、抗拒疾、抗心律失常、抗血栓等作用。因此,这类化合物在医药学、农业化学、材料科学等领域中存在广泛的应用,其合成与发展是合成化学、药物化学和材料化学研究领域的重要课题。近年来,此研究领域得到了快速的发展,一系列具有重要实践价值的异喹啉二酮合成方法相续被报道。
3.目前,制备异喹啉酮类化合物主要通过加入过渡金属催化或者介导的合成方法。在众多过渡金属参与的异喹啉酮合成法中,常用的催化剂包括钯、铜、钌和铑等金属以及各种配体,通过碳氢、碳氮键的活化与构建或者以自由基方式实现喹啉环的合成,这些方法大多数简洁高效,底物适用性较广,但也存在一些不足之处:1)一些催化剂和配体价格不菲,而且制备过程繁琐;2)过渡金属毒性较大且难以彻底从产物中除去;3)反应温度高,副产物多。
4.传统的氧化还原反应都需要等当量或过量的氧化还原试剂,同时常常伴随着高温。有机电化学在十九世纪就已经开始建立,但在二十世纪六十年代才开始出现对其机理的研究。近年来绿色化学的兴起导致有机光化学的复兴接着带动自由基化学的复兴。有机电化学同样可以发生单电子转移产生自由基,并且相较于光化学有其更加独特的优点。这种环保的策略近几年正在飞速地发展并逐渐改变了有机合成方式,无可厚非地成为绿色化学中最重要的部分之一,许多的化学家们都在致力于研究它的机理和开发它的进一步应用。因此,本发明提供了一种利用微通道反应装置连续制备烷基化异喹啉酮类化合物的方法。
技术实现要素:
5.发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种利用微通道反应装置连续制备烷基化异喹啉酮类化合物的方法。
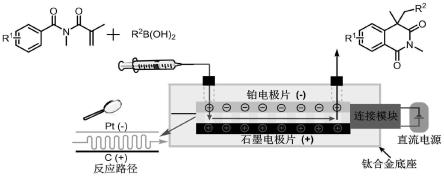
6.为了解决上述技术问题,本发明公开了一种利用微通道反应装置连续制备烷基化异喹啉酮类化合物的方法,如图2所示,将含式1所示的n-甲基丙烯酰基-n-甲基苯甲酰胺类化合物、式2所示的烷基硼酸、电解质和溶剂的混合溶液泵入设有电极的微通道反应装置中的微通道反应器中,进行连续电解反应,即得含有式3所示的烷基化异喹啉酮类化合物的反应液;
[0007][0008]
其中,
[0009]
r1选自氢、4-甲基、4-甲氧基、4-氯、4-氟或2-甲基,优选为氢、4-甲基、4-甲氧基或4-氯,进一步优选为氢、4-甲基或4-甲氧基;
[0010]
r2选自环己基、异丙基或正丁基,优选为环己基。
[0011]
其中,所述混合溶液中式1所示的n-甲基丙烯酰基-n-甲基苯甲酰胺类化合物的浓度为0.02~0.06mmol/ml,优选为0.03mmol/ml。
[0012]
其中,所述混合溶液中式2所示的烷基硼酸的浓度为0.02~0.12mmol/ml,优选为0.04~0.18mmol/ml,进一步优选为0.06mmol/ml。
[0013]
其中,所述混合溶液中电解质的浓度为0.02~0.12mmol/ml,优选为0.04~0.18mmol/ml,进一步优选为0.06mmol/ml。
[0014]
其中,所述电解质为高氯酸锂、四丁基六氟磷酸铵、四丁基四氟硼酸铵、四丁基碘化铵、四乙基高氯酸铵和碘化钠中的任意一种或几种组合,优选高氯酸锂。
[0015]
其中,所述溶剂为乙腈、二氯甲烷、乙酸和甲醇中的任意一种或几种组合,优选为乙腈和乙酸的混合溶剂,进一步优选为乙腈与乙酸体积比为4:1的混合溶剂。
[0016]
其中,所述设有电极的微通道反应装置包括泵、阴极片、阳极片、微通道反应器和接收器;所述泵、微通道反应器和接收器通过管道依次串联,所述微通道反应器的两侧分别设有阴极片和阳极片。
[0017]
其中,所述阳极片为石墨板,所述阴极片为铂片。
[0018]
其中,所述微通道反应器的反应体积为125~400μl。
[0019]
其中,所述混合溶液泵入泵入微通道反应器的流速为45~459μl/min,优选为112.5μl/min。
[0020]
其中,所述反应的电流强度为5~20ma,优选为8~12ma,进一步优选为10ma。
[0021]
其中,所述反应的温度为5~35℃,优选为20~30℃,进一步优选为室温。
[0022]
其中,所述反应的停留时间为0.5~5min,优选为1~3min,进一步优选为2min。
[0023]
有益效果:与现有技术相比,本发明具有如下优势:
[0024]
(1)本发明首次报道了通过电化学氧化制备烷基化喹啉酮化合物,克服了传统氧化体系的反应流程周期长、需要昂贵催化剂的问题,该方法有利于放大反应,并且反应过程安全、高效、绿色。
[0025]
(2)本发明采用微通道反应装置,反应时间短,产物收率高,显著的提高了反应效率。
[0026]
(3)本发明无需添加昂贵的有机催化剂或金属催化剂,操作简便,绿色高效,成本低。
[0027]
(4)本发明通过注射泵及微通道反应装置连续反应,制备工艺易操作控制,反应条件温和,安全性高,具有更好的工业放大潜力。
附图说明
[0028]
下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
[0029]
图1是本发明微通道电合成反应装置图。
[0030]
图2为本发明的反应路径图。
具体实施方式
[0031]
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0032]
下述实施例利用图1所述的微通道反应装置,按照下述步骤:(1)将按比例配置好的均相溶液添加到注射泵中;(2)通过注射泵按照一定比例注入到微通道反应装置中进行混合和反应;(3)调节所需电流;(4)收集流出反应液,通过高效液相测得反应转化收率,再经柱层析(石油醚:乙酸乙酯=5:1)分离得到目标产物,计算分离收率。
[0033]
其中,如表1所示的烷基化异喹啉酮类化合物均为通过本发明方法合成得到的产物,且经过核磁表征确证。
[0034]
其中,如表2所示的为本发明的反应物。
[0035]
表1本发明的烷基化异喹啉酮类化合物
[0036]
[0037][0038]
表2本发明的反应物
[0039][0040]
实施例1化合物3a的合成:
[0041]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸2a和0.6mmol高氯酸锂(0.064g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为91%,柱层析分离后得到产物3a,收率为84%。
[0042]1h nmr(400mhz,chloroform-d)δ8.14(d,j=7.8hz,1h),7.58
–
7.52(m,1h),7.31
–
7.24(m,2h),3.33(d,j=1.2hz,3h),2.23(dd,j=14.0,7.3hz,1h),1.81(dd,j=14.1,4.5hz,1h),1.49(s,3h),1.34
–
1.28(m,3h),1.18
–
1.12(m,2h),0.85
–
0.78(m,4h),0.72
–
0.66(m,2h).
[0043]
13
c nmr(101mhz,chloroform-d)δ176.8,164.5,143.9,133.7,128.8,127.1,125.7,124.5,49.5,46.7,34.7,34.5,33.1,31.4,27.1,26.1,25.9,25.8.
[0044]
实施例2化合物3a的合成:
[0045]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol四丁基四氟硼酸铵(0.198g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为85%,柱层析分离后得到产物3a,收率为73%。
[0046]
实施例3化合物3a的合成:
[0047]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol四丁基六氟磷酸铵(0.232g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为89%,柱层析分离后得到产物3a,收率为76%。
[0048]
实施例4化合物3a的合成:
[0049]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于乙腈(10ml)中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为80%,柱层析分离后得到产物3a,收率为63%。
[0050]
实施例5化合物3a的合成:
[0051]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于二氯甲烷(10ml)中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为76%,柱层析分离后得到产物3a,收率为54%。
[0052]
实施例6化合物3a的合成:
[0053]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于甲醇(10ml)中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为77%,柱层析分离后得到产物3a,收率为58%。
[0054]
实施例7化合物3a的合成:
[0055]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为225μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间
1min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为84%,柱层析分离后得到产物3a,收率为75%。
[0056]
实施例8化合物3a的合成:
[0057]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为56μl/min;施加电流为10ma;微通道反应器反应体积v=225μl,反应时间4min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为80%,柱层析分离后得到产物3a,收率为62%。
[0058]
实施例9化合物3a的合成:
[0059]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为5ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为81%,柱层析分离后得到产物3a,收率为64%。
[0060]
实施例10化合物3a的合成:
[0061]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为112.5μl/min;施加电流为20ma;微通道反应器反应体积v=225μl,反应时间2min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为86%,柱层析分离后得到产物3a,收率为73%。
[0062]
实施例11化合物3a的合成:
[0063]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为125μl/min;施加电流为10ma;微通道反应器反应体积v=125μl,反应时间1min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为81%,柱层析分离后得到产物3a,收率为69%。
[0064]
实施例12化合物3a的合成:
[0065]
将0.3mmol(0.052g)化合物1a,0.6mmol(0.077g)环已基硼酸和0.6mmol高氯酸锂(0.064g)溶于乙腈/乙酸(4:1,10ml)的混合溶剂中,得到均相溶液,添加到注射泵a中;注射泵a的注射流速为400μl/min;施加电流为10ma;微通道反应器反应体积v=400μl,反应时间1min;在微通道反应器反应历经一个周期后,收集反应液体,hplc的方法计算反应转化收率为90%,柱层析分离后得到产物3a,收率为77%。
[0066]
实施例13化合物3b的合成:
[0067]
与实施例1方法相同,所不同的是,参与反应的是化合物1b,收率为83%,柱层析分离后得到产物3b。
[0068]1h nmr(400mhz,chloroform-d)δ8.11(d,j=7.9hz,1h),7.28
–
7.22(m,1h),7.16(s,1h),3.33(s,3h),2.45(s,3h),2.27(dd,j=13.0,7.4hz,1h),1.86(dd,j=13.0,4.8hz,1h),1.53(s,3h),1.43
–
1.35(m,3h),1.24
–
1.22(m,1h),1.11
–
1.02(m,1h),0.95
–
0.91(m,4h),0.77
–
0.74(m,2h);
[0069]
13
c nmr(101mhz,chloroform-d)δ177.2,164.5,144.5,143.7,128.8,128.1,126.0,122.0,77.3,77.0,76.7,49.6,46.6,34.8,34.2,33.0,31.3,27.0,26.2,26.0,25.7,22.3.
[0070]
实施例14化合物3c的合成:
[0071]
与实施例1方法相同,所不同的是,参与反应的是化合物1c,收率为84%,柱层析分离后得到产物3c。
[0072]1h nmr(400mhz,chloroform-d)δ8.21(d,j=8.4hz,1h),6.97(dd,j=8.3,2.5hz,1h),6.82(d,j=2.4hz,1h),3.91(s,3h),3.37(s,3h),2.32(dd,j=14.0,7.5hz,1h),1.81(dd,j=14.0,4.8hz,1h),1.55(s,3h),1.48
–
1.30(m,3h),1.22
–
1.19(m,2h),0.97
–
0.92(m,4h),0.79
–
0.73(m,2h);
[0073]
13
c nmr(101mhz,chloroform-d)δ176.6,164.0,163.5,146.1,131.2,117.6,113.0,110.6,55.5,49.9,46.8,34.6,34.3,33.2,31.8,27.0,26.3,26.0,25.9.
[0074]
实施例15化合物3d的合成:
[0075]
与实施例1方法相同,所不同的是,参与反应的是化合物1d,收率为77%,柱层析分离后得到产物3d。
[0076]1h nmr(400mhz,chloroform-d)δ8.16(d,j=8.3hz,1h),7.37
–
7.36(m,2h),3.34(s,3h),2.32(dd,j=14.2,7.4hz,1h),1.81(dd,j=14.3,4.2hz,1h),1.55(s,3h),1.43
–
1.30(m,3h),1.16(dd,j=14.9,12.6hz,2h),0.97
–
0.93(m,4h),0.85
–
0.77(m,2h);
[0077]
13
c nmr(101mhz,chloroform-d)δ176.2,163.6,145.7,140.2,130.5,127.9,125.9,123.0,49.5,46.3,34.8,34.2,32.9,31.5,27.2,26.0,25.9,25.6.
[0078]
实施例16化合物3e的合成:
[0079]
与实施例1方法相同,所不同的是,参与反应的是化合物1e,收率为63%,柱层析分离后得到产物3b。
[0080]1h nmr(400mhz,chloroform-d)δ7.44(t,j=7.2hz,1h),7.29(d,j=7.7hz,1h),7.21(d,j=7.5hz,1h),3.33(s,3h),2.79(s,3h),2.30(dd,j=14.2,7.3hz,1h),1.88(dd,j=14.1,4.7hz,1h),1.52(s,3h),1.49
–
1.30(m,3h),1.15
–
1.10(m,2h),0.93
–
0.88(m,4h),0.73
–
0.71(m,2h);
[0081]
13
c nmr(101mhz,chloroform-d)δ176.4,165.2,145.2,142.4,132.3,131.1,124.2,122.9,50.4,46.6,34.2,34.3,33.0,31.5,27.1,26.4,26.1,26.0,24.1.
[0082]
实施例17化合物3f的合成:
[0083]
与实施例1方法相同,所不同的是,参与反应的是化合物2b,收率为77%,柱层析分离后得到产物3f。
[0084]1h nmr(400mhz,chloroform-d)δ8.26(d,j=7.3hz,1h),7.65-7.61(m,1h),7.44-7.40(m,2h),3.39(s,3h),2.36-2.27(q,1h),1.97-1.92(q,1h),1.58(s,3h),1.22-1.11(m,1h),0.63(d,j=6.6hz,3h),0.61(d,j=6.6hz,3h);
[0085]
13
c nmr(101mhz,chloroform-d)δ176.9,164.3,143.9,133.8,128.5,127.2,126.6,124.6,50.7,46.9,31.6,27.2,25.4,23.9,22.2.
[0086]
实施例18化合物3g的合成:
[0087]
与实施例1方法相同,所不同的是,参与反应的是化合物2c,收率为74%,柱层析分
离后得到产物3g。
[0088]1h nmr(400mhz,chloroform-d)δ8.25(d,j=7.4hz,1h),7.66(t,j=7.8hz,1h),7.47-7.42(m,2h),3.40(s,3h),2.32-2.24(m,1h),1.89-1.84(m,1h),1.63(s,3h),1.21-1.05(m,4h),0.92-0.85(m,1h),0.72-0.67(m,4h);
[0089]
13
c nmr(101mhz,chloroform-d)δ176.8,164.3,143.8,133.5,128.7,127.1,125.2,124.9,47.8,43.3,31.7,29.1,27.4,24.8,22.3,13.9.
[0090]
本发明提供了一种利用电化学微通道反应装置连续制备烷基化异喹啉酮类化合物的方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1