一种利用氟利昂型甲烷作为碳一合成子合成标记卤代吲哚酮的方法及其应用
1.本发明涉及有机化学合成技术领域,具体涉及一种利用氟利昂型甲烷作为碳一合成子合成标记吲哚类生物碱的方法及其应用。
背景技术:
2.吲哚类生物碱是一类重要的具有生物药物活性的分子。其中通过丙烯酰胺的自由基串联环化过程合成吲哚酮的方法可以方便地引入吲哚结构中不同位置的各类取代基。由于有机卤代物常常表现出各类生物药物活性,且卤原子容易发生官能团转化,因此卤代吲哚酮的合成受到了人们的重视。
3.但是目前的方法中常常利用低沸点的二氯甲烷等溶剂,通过攫氢形成碳自由基参与反应。反应通常需要在远高于溶剂沸点的温度进行,同时反应中需要加入化学当量的氧化剂、攫氢试剂和碱,使得反应具有毒性和环境不友好性(org.chem.front.2014,1,1289;org.lett.2014,16,4698)。反应中引入的卤代烷烃主要限制于氯代甲烷,对氟、溴、碘等卤素以及氘等同位素也不能实现有效的引入。有机电化学反应避免了传统氧化剂和还原剂的使用,能够方便地产生自由基等活性物种,是一种高效绿色、富有前景的合成方法(j.am.chem.soc.2020,142,20661)。
4.稳定同位素标记(如2h,
13
c)等在同位素示踪、药物分子的药代动力学研究和改善等方面具有广泛的应用。目前以前稳定同位素标记的引入多是依靠氢氘交换、氘代甲基的引入等方法,这造成可氘代的位点受限、合成方法复杂,通过碳一合成子在分子骨架上引入氘代标记的方法报道较少(j.med.chem.2019,62,5276)。
技术实现要素:
5.本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种利用氟利昂型甲烷作为碳一合成子合成标记卤代吲哚酮的方法及其应用。
6.本发明的目的可以通过以下技术方案来实现:
7.一种利用氟利昂型甲烷作为碳一合成子合成卤代吲哚酮的方法,该方法为:在电化学反应系统中,将底物丙烯酰胺、氟利昂型甲烷和电解质溶于溶剂中,利用氟利昂型甲烷上碳卤键的异裂生成的卤素离子和碳自由基,随后碳自由基被丙烯酰胺捕获并发生自由基环化反应,生成卤代吲哚酮。
8.进一步地,所述氟利昂型卤甲烷包括一卤、二卤、三卤或四卤,包含有氟、氯、溴或碘原子的取代甲烷。
9.进一步地,所述丙烯酰胺的化学结构式为:
[0010][0011]
其中,r1选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0012]
r2选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0013]
r3选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0014]
r4选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基。
[0015]
进一步地,所述的电解质包括四丁基四氟硼酸铵、四甲基硼酸铵、四丁基六氟磷酸铵或高氯酸锂。
[0016]
进一步地,所述的溶剂包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜或水,优选n,n-二甲基甲酰胺。
[0017]
进一步地,所述的电化学反应系统中,使用石墨毡作为正极、泡沫镍作为负极,在80-120℃、氩气氛围、4-6ma恒定电流条件下,反应6-12h,其中,反应时间按所添加丙烯酰胺完全转化所需要电子的量的1.5倍进行计算,丙烯酰胺浓度范围为0.02-0.06mol/l。
[0018]
一种如上所述方法合成的卤代吲哚酮,该卤代吲哚酮的化学结构式为:
[0019][0020]
其中,r1选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0021]
r2选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0022]
r3选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0023]
r4选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0024]
rx选自包含有氟、氯、溴、碘卤原子和2h,3h,
13
c,
14
c同位素的二卤代、三卤代甲基。
[0025]
一种如上所述卤代吲哚酮的应用,该卤代吲哚酮用于合成同位素标记或未标记的吲哚类生物碱。
[0026]
可以采用以下制备路线:
[0027][0028][0029]
进一步地,引入的标记包括2h,3h,
13
c或
14
c。
[0030]
进一步地,所述的吲哚类生物碱包括(
±
)-physostigmine、(
±
)-esermethole或(
±
)-lansai b。
[0031]
毒扁豆碱(physostigmine)是一种从非洲出产的毒扁豆种子中提出的吲哚类生物碱,易透过血脑屏障,具有可逆性抑制胆碱酯酶的作用,在临床上用于缩瞳、降低眼压和逆转抗胆碱能药物所致中枢毒性作用。esermethole和lansai b是从天然植物和菌群中分离所得的吲哚类生物碱,表现出抗癌等生物药物活性。
[0032]
对药物分子进行各种标记是现代医药学的重要组成部分,常见的标记包括氢同位素(2h,3h)、碳同位素(
13
c,
14
c)、氟同位素(
18
f)等。研究表明,稳定同位素标记(如氘代)可以对药物分子的药代动力学等性质产生影响,从而影响药物效果;稳定同位素示踪已经成为生物药物化学研究中重要的分析工具。
[0033]
本发明技术采用经济环保的电化学反应实现了以氟利昂型甲烷作为碳一合成子,通过碳卤键还原断裂及后续的自由基串联环化过程实现了各类不同卤代的吲哚酮化合物的合成。所得的卤代吲哚酮可进一步转化为各类同位素标记或未标记的吲哚类生物碱,包括(
±
)-physostigmine、(
±
)-esermethole、(
±
)-lansai b,具有较高的应用价值。
[0034]
与现有技术相比,本发明具有以下优点:
[0035]
(1)本发明方法采用绿色环保的电化学反应系统,无需添加常规氧化剂或还原剂、金属催化剂和碱,无需使用低沸点的卤甲烷在高温下反应,反应条件安全、高效,减少了对环境的污染;
[0036]
(2)本发明方法以氟利昂型甲烷作为碳一合成子,实现了卤代烷烃碳卤键的断裂及对烷基自由基的有效捕获,所得的卤代吲哚酮可进一步转化为一系列具有生物药物活性的吲哚类生物碱;
[0037]
(3)本发明方法的底物丙烯酰胺及氟利昂型甲烷具有多样性,可以构建多类不同卤代的吲哚酮化合物;
[0038]
(4)本发明方法可以方便地由氟利昂型甲烷向卤代吲哚酮中引入氟、氢同位素、碳同位素等标记,进而可以合成标记的吲哚类生物碱分子。
具体实施方式
[0039]
下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
[0040]
一种利用氟利昂型甲烷作为碳一合成子合成卤代吲哚酮的方法,该方法为:在电化学反应系统中,将底物丙烯酰胺、氟利昂型甲烷和电解质溶于溶剂中,利用氟利昂型甲烷上碳卤键的异裂生成的卤素离子和碳自由基,随后碳自由基被丙烯酰胺捕获并发生自由基环化反应,生成卤代吲哚酮,该卤代吲哚酮的化学结构式为:
[0041][0042]
其中,r1选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0043]
r2选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0044]
r3选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0045]
r4选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基
的杂环基或卤素取代基;
[0046]
rx选自包含有氟、氯、溴、碘卤原子和2h,3h,
13
c,
14
c同位素的二卤代、三卤代甲基。
[0047]
得到的卤代吲哚酮经过多步化学转化,可以制得到一系列同位素标记或未标记的吲哚类生物碱,包括(
±
)-physostigmine、(
±
)-esermethole、(
±
)-lansai b,具有较高的应用价值。
[0048]
其中,氟利昂型卤甲烷包括一卤、二卤、三卤或四卤,包含有氟、氯、溴或碘原子的取代甲烷。丙烯酰胺的化学结构式为:
[0049][0050]
其中,r1选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0051]
r2选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0052]
r3选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基;
[0053]
r4选自h、烷基、支链烷基、环烷基、芳香基、含取代基的芳香基、杂环基、含取代基的杂环基或卤素取代基。
[0054]
电解质包括四丁基四氟硼酸铵、四甲基硼酸铵、四丁基六氟磷酸铵或高氯酸锂。溶剂包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜或水,优选n,n-二甲基甲酰胺。
[0055]
电化学反应系统中,使用石墨毡作为正极、泡沫镍作为负极,在80-120℃、氩气氛围、4-6ma恒定电流条件下,反应6-12h,其中,反应时间按所添加丙烯酰胺完全转化所需要电子的量的1.5倍进行计算,丙烯酰胺浓度范围为0.02-0.06mol/l。
[0056]
本实施方式中,化合物的氢核磁共振谱(1h nmr)由bruker avance iii hd 400或bruker avance iii hd 500测定;质谱(esi-ms)由waters acquitytm uplc&q-tof ms premier测定;所用试剂均为市售试剂。
[0057]
本发明的合成方法可以制备如下所示的卤代吲哚酮化合物:
[0058][0059]
实施例1
[0060]
卤代吲哚酮化合物(i-1)的制备
[0061]
将0.2mmol n-benzyl-4-hydroxy-n-(4-methoxyphenyl)-2-methylene-butanamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到浅黄色油状液体(i-1),收率为49%。
[0062]
化合物(i-1)为:
[0063]1h nmr(400mhz,cdcl3)δ7.34
–
7.27(m,5h),6.79(d,j=2.4hz,1h),6.75
–
6.72(m,1h),6.69
–
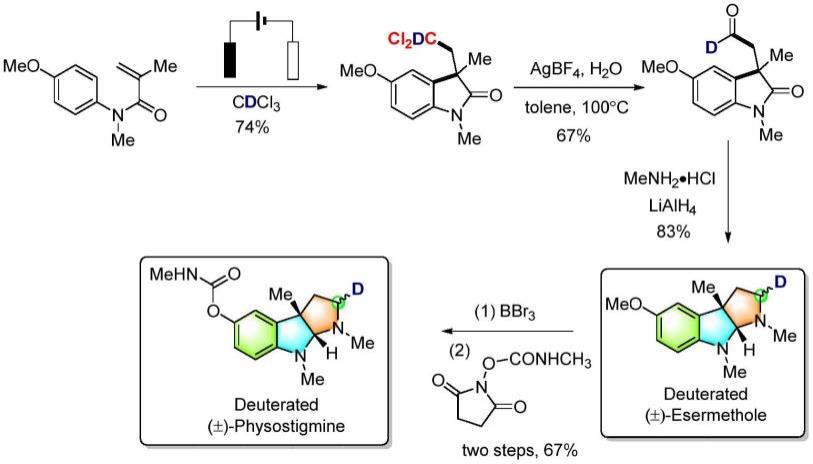
6.67(m,1h),5.44(dd,j=8.8,4.4hz,1h),5.01(d,j=15.6hz,1h),4.72(d,j=15.6hz,1h),3.60
–
3.54(m,1h),3.51
–
3.45(m,1h),3.13(dd,j=14.8,8.8hz,1h),2.76(dd,j=14.8,4.4hz,1h),2.28
–
2.21(m,1h),2.06
–
1.99(m,1h).
[0064]
13
c nmr(101mhz,cdcl3)δ178.6,156.2,136.6,135.6,130.6,128.8,127.7,127.6,112.6,110.8,110.2,69.2,58.5,55.8,50.3,49.8,44.5,41.6.
[0065]
hrms(esi)m/z calculated for c
20h21
cl2nnao3[m+na
+
]416.0791,found 416.0785.
[0066]
实施例2:
[0067]
卤代吲哚酮化合物(i-2)的制备
[0068]
将0.2mmol n-benzyl-n-(4-bromophenyl)-5-methyl-2-methylenehex-4-enamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到浅黄色油状液体(i-2),收率为56%。
[0069]
化合物(i-2)为:
[0070]1h nmr(400mhz,cdcl3)δ7.31
–
7.24(m,7h),6.57(d,j=8.8hz,1h),5.46(dd,j=9.0,4.6hz,1h),5.14(d,j=15.8hz,1h),4.79
–
4.76(m,1h),4.58(d,j=15.7hz,1h),3.08(dd,j=14.8,9.0hz,1h),2.77(dd,j=14.8,4.6hz,1h),2.60(dd,j=13.8,8.3hz,1h),2.47(dd,j=13.7,7.0hz,1h),1.59(s,3h),1.50(s,3h).
[0071]
13
c nmr(101mhz,cdcl3)δ177.7,142.4,137.4,135.2,131.5,131.3,128.8,127.7,127.3,126.4,116.1,115.1,110.9,69.5,51.9,48.7,44.1,37.8,25.9,18.0.
[0072]
hrms(esi)m/z calculated for c
22h22
brcl2nnao[m+na
+
]488.0154,found 488.0149.
[0073]
实施例3:
[0074]
卤代吲哚酮化合物(i-3)的制备
[0075]
将0.2mmol methyl 3-(benzyl(4-fluorophenyl)carbamoyl)but-3-enoate、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到浅黄色固体(i-3),收率为59%。
[0076]
化合物(i-3)为:
[0077]1h nmr(400mhz,cdcl3)δ7.37
–
7.28(m,5h),7.07(dd,j=7.8,2.6hz,1h),6.91(td,j=8.8,2.6hz,1h),6.68(dd,j=8.6,4.2hz,1h),5.51(dd,j=7.8,5.2hz,1h),5.01(d,j=15.6hz,1h),4.81(d,j=15.7hz,1h),3.51(s,3h),3.03
–
2.90(m,4h).
[0078]
13
c nmr(101mhz,cdcl3)δ177.2,169.3,159.1(d,j=241.9hz),139.5(d,j=2.1hz),135.2,130.1(d,j=8.0hz),128.8,127.8,127.5,115.3(d,j=23.2hz),111.8(d,j=25.0hz),110.3(d,j=8.1hz),68.6,51.9,49.0,48.9(d,j=1.8hz),44.6,41.4.
[0079]
hrms(esi)m/z calculated for c
20h18
cl2fnnao3[m+na
+
]432.0540,found 432.0539.
[0080]
实施例4:
[0081]
卤代吲哚酮化合物(i-4)的制备
[0082]
将0.2mmol methyl 3-(1,2,3,4-tetrahydroquinoline-1-carbonyl)but-3-enoate、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到浅黄色油状液体(i-4),收率为67%。
[0083]
化合物(i-4)为:
[0084]1h nmr(400mhz,cdcl3)δ7.10
–
7.06(m,2h),6.97
–
6.93(m,1h),5.42(t,j=6.5hz,1h),3.75
–
3.65(m,2h),3.51(s,3h),2.94(d,j=6.5hz,2h),2.91
–
2.81(m,2h),2.77(t,j=6.1hz,2h),2.04
–
1.93(m,2h).
[0085]
13
c nmr(101mhz,cdcl3)δ176.2,169.7,140.0,127.9,126.6,122.1,121.3,120.7,69.3,51.7,49.6,48.6,41.1,39.1,24.6,20.9.
[0086]
hrms(esi)m/z calculated for c
16h17
cl2nnao3[m+na
+
]364.0478,found 364.0478.
[0087]
实施例5:
[0088]
卤代吲哚酮化合物(i-5)的制备
[0089]
将0.2mmol 1-(3,4-dihydroquinolin-1(2h)-yl)-5-methyl-2-methylenehex-4-en-1-one、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到无色油状液体(i-5),收率为74%。
[0090]
化合物(i-5)为:
[0091]1h nmr(400mhz,cdcl3)δ7.06
–
7.04(m,1h),7.01
–
7.00(m,1h),6.97
–
6.93(m,1h),5.37(dd,j=9.6,3.9hz,1h),4.93
–
4.89(m,1h),3.73
–
3.62(m,2h),2.99(dd,j=14.8,9.6hz,1h),2.79
–
2.73(m,3h),2.48(dd,j=14.0,7.7hz,1h),2.36(dd,j=14.0,7.5hz,
1h),1.99
–
1.93(m,2h),1.62(s,3h),1.50(s,3h).
[0092]
13
c nmr(101mhz,cdcl3)δ177.3,139.8,136.6,127.6,127.3,121.7,121.3,120.4,116.8,70.2,52.8,48.0,39.0,36.9,25.9,24.7,21.1,18.1.
[0093]
hrms(esi)m/z calculated for c
18h21
cl2nnao[m+na
+
]360.0892,found 360.0890.
[0094]
实施例6:
[0095]
卤代吲哚酮化合物(i-6)的制备
[0096]
将0.2mmol n-benzyl-2-(2-methoxybenzyl)-n-phenylacrylamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到无色膏状物(i-6),收率为67%。
[0097]
化合物(i-6)为:
[0098]1h nmr(400mhz,cdcl3)δ7.23
–
7.21(m,3h),7.18
–
7.14(m,1h),7.11
–
7.07(m,2h),7.00
–
6.96(m,4h),6.75(td,j=7.4,0.9hz,1h),6.65(d,j=8.2hz,1h),6.52
–
6.50(m,1h),5.41(dd,j=9.4,4.1hz,1h),4.99(d,j=15.8hz,1h),4.61(d,j=15.9hz,1h),3.49(s,3h),3.43(d,j=13.0hz,1h),3.26(dd,j=14.8,9.4hz,1h),3.04(d,j=13.0hz,1h),2.91(dd,j=14.8,4.1hz,1h).
[0099]
13
c nmr(101mhz,cdcl3)δ178.1,157.5,143.1,135.5,131.9,128.5,128.3,128.2,127.2,127.1,124.5,123.3,121.5,119.9,110.1,109.1,69.9,54.6,52.8,48.9,43.9,37.7.
[0100]
hrms(esi)m/z calculated for c
25h23
cl2nnao2[m+na
+
]462.0998,found 462.0996.
[0101]
实施例7:
[0102]
卤代吲哚酮化合物(i-7)的制备
[0103]
将0.2mmol(e)-n,2-dimethyl-n-phenylbut-2-enamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到无色油状物(i-7,一对非对映异构体),收率为68%。
[0104]
化合物(i-7)为:
[0105]1h nmr(400mhz,cdcl3)δ7.34
–
7.26(m,1h),7.21(d,j=7.1hz,1h),7.11
–
7.03(m,1h),6.88
–
6.84(m,1h),5.77(d,j=2.8hz,0.19h),5.65(d,j=1.6hz,0.77h),3.24(s,0.6h),3.17(s,2.4h),2.82
–
2.76(m,0.2h),2.71
–
2.66(m,0.8h),1.54
–
1.51(m,3h),1.45
–
1.42(m,3h).
[0106]
13
c nmr(101mhz,cdcl3)δ179.3,178.0,143.2,131.9,131.0,128.5,128.2,124.3,122.8,122.6,122.5,108.4,108.3,75.3,75.0,51.6,50.6,50.0,49.5,26.3,26.2,23.5,22.8,9.0,7.5.
[0107]
hrms(esi)m/z calculated for c
13h15
cl2nnao[m+na
+
]294.0423,found 294.0421.
[0108]
实施例8:
[0109]
卤代吲哚酮化合物(i-8)的制备
[0110]
将0.2mmol n-methyl-n-phenylcyclohex-1-ene-1-carboxamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol溴二氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到浅黄色固体(i-8,一对非对映异构体),收率为41%。
[0111]
化合物(i-8)为:
[0112]
上点(浅黄色固体):
[0113]1h nmr(400mhz,cdcl3)δ7.33
–
7.28(m,1h),7.13
–
7.07(m,2h),6.84(d,j=7.8hz,1h),5.35(s,1h),3.15(s,3h),2.54
–
2.46(m,2h),2.43
–
2.31(m,1h),2.14
–
2.11(m,2h),1.82
–
1.73(m,2h),1.60
–
1.47(m,2h).
[0114]
13
c nmr(101mhz,cdcl3)δ177.1,143.3,131.8,128.5,122.7,121.6,108.1,73.9,55.8,49.3,36.6,26.1,25.5,20.1,19.3.
[0115]
hrms(esi)m/z calculated for c
15h17
cl2nnao[m+na
+
]320.0579,found 320.0580.
[0116]
下点(浅黄色固体):
[0117]1h nmr(400mhz,cdcl3)δ7.51(d,j=7.5hz,1h),7.33(t,j=7.8hz,1h),7.04(t,j=7.4hz,1h),6.90(d,j=7.8hz,1h),5.24(d,j=6.2hz,1h),3.23(s,3h),2.83
–
2.77(m,1h),2.41
–
2.36(m,1h),2.11
–
2.08(m,1h),1.95
–
1.88(m,1h),1.85
–
1.65(m,4h),1.48
–
1.42
(m,1h).
[0118]
13
c nmr(101mhz,cdcl3)δ179.6,143.4,130.0,128.2,125.7,121.8,108.6,75.1,51.5,51.5,36.6,26.6,24.9,24.6,20.9.
[0119]
hrms(esi)m/z calculated for c
15h17
cl2nnao[m+na
+
]320.0579,found 320.0581.
[0120]
实施例9:
[0121]
卤代吲哚酮化合物(i-9)的制备
[0122]
将0.2mmol n-methyl-n-phenylmethacrylamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol氘代氯仿溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到无色油状物(i-9),收率为83%。
[0123]
化合物(i-9)为:
[0124]1h nmr(400mhz,cdcl3)δ7.31(td,j=7.7,1.2hz,1h),7.20
–
7.18(m,1h),7.11
–
7.07(m,1h),6.87(d,j=7.8hz,1h),3.20(s,3h),3.02(d,j=14.8hz,1h),2.69(d,j=14.8hz,1h),1.39(s,3h).
[0125]
13
c nmr(101mhz,cdcl3)δ178.9 143.3,131.0,128.6,122.6,122.6,108.6,69.4(j=27.3hz),49.9,47.1,26.4,25.4.
[0126]
hrms(esi)m/z calculated for c
12h12
cl2dnnao[m+na
+
]281.0329,found 281.0327.
[0127]
实施例10:
[0128]
卤代吲哚酮化合物(i-10)的制备
[0129]
将0.2mmol n-methyl-n-phenylmethacrylamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol三溴甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到无色油状物(i-10),收率为64%。
[0130]
化合物(i-10)为:
[0131]1h nmr(400mhz,cdcl3)δ7.32(td,j=7.7,1.3hz,1h),7.19
–
7.17(m,1h),7.12
–
7.08(m,1h),6.87(d,j=7.8hz,1h),5.30(dd,j=9.6,4.3hz,1h),3.27(dd,j=15.1,9.6hz,1h),3.20(s,3h),2.99(dd,j=15.2,4.3hz,1h),1.37(s,3h).
[0132]
13
c nmr(101mhz,cdcl3)δ178.8,143.5,130.7,128.6,122.6,108.6,51.5,48.5,39.6,26.5,25.6.
[0133]
hrms(esi)m/z calculated for c
12h13
br2nnao[m+na
+
]367.9256,found 367.9254.
[0134]
实施例11:
[0135]
卤代吲哚酮化合物(i-11)的制备
[0136]
将0.2mmol n-methyl-n-phenylmethacrylamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol二溴氯甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到无色油状物(i-11,一对非对映异构体),收率为66%。
[0137]
化合物(i-11)为:
[0138]1h nmr(400mhz,cdcl3)δ7.32(tdd,j=7.7,3.8,1.1hz,1h),7.20
–
7.18(m,1h),7.12
–
7.08(m,1h),6.87(d,j=7.8hz,1h),5.43
–
5.36(m,1h),3.21
–
3.09(m,4h),2.87
–
2.81(m,1h),1.39(s,1.3h),1.38(s,1.5h).
[0139]
13
c nmr(101mhz,cdcl3)δ179.0,178.7,143.5,143.4,131.1,130.7,128.6,128.5,122.7,122.6,122.6,108.6,108.6,55.7,55.6,51.1,50.8,48.0,47.8,26.4,25.6,25.3.
[0140]
hrms(esi)m/z calculated for c
12h13
brclnnao[m+na
+
]323.9761,found 323.9762.
[0141]
实施例12:
[0142]
卤代吲哚酮化合物(i-12)的制备
[0143]
将0.2mmol n-methyl-n-phenylmethacrylamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol二溴二氟甲烷溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到无色油状物(i-12),收率为38%。
[0144]
化合物(i-12)为:
[0145]1h nmr(400mhz,cdcl3)δ7.31(td,j=7.7,1.1hz,1h),7.27
–
7.25(m,1h),7.08(td,j=7.5,0.7hz,1h),6.88(d,j=7.8hz,1h),3.34
–
3.22(m,4h),3.10
–
2.98(m,1h),1.38(s,
3h).
[0146]
13
c nmr(101mhz,cdcl3)δ178.3,142.8,130.8,128.4,123.8(t,j=1.7hz),122.5,119.5(t,j=307.5hz),108.4,50.9(t,j=21.0hz),45.9,26.4,25.3.
[0147]
19
f nmr(376mhz,cdcl3)δppm-39.9(d,j=155.0hz,1f),-43.9(d,j=154.9hz,1f).
[0148]
hrms(esi)m/z calculated for c
12h12
brf2nnao[m+na
+
]325.9963,found 325.9963.
[0149]
实施例13:
[0150]
氘代(
±
)-esermethole(i-13)的制备
[0151]
将0.2mmol n-(4-methoxyphenyl)-n-methylmethacrylamide、0.4mmol四丁基四氟硼酸铵加入10ml史莱克管中,使用石墨毡作为阳极、铂片作为阴极,用导线穿过的橡皮塞塞住密封,抽真空并置换氩气,反复三次。将2mmol氘代氯仿溶于4ml n,n-二甲基甲酰胺中,用注射器将溶液打入反应管内,在恒定电流(5ma)、100摄氏度、搅拌条件下反应12h,对反应液使用乙酸乙酯进行萃取,对所得的有机相进行浓缩、柱层析得到白色固体(i),收率为74%。
[0152]
化合物(i)为:
[0153]1h nmr(400mhz,cdcl3)δ6.83
–
6.75(m,3h),3.79(s,3h),3.16(s,3h),2.99(d,j=14.8hz,1h),2.65(d,j=14.8hz,1h),1.36(s,3h).
[0154]
13
c nmr(101mhz,cdcl3)δ178.5,156.0,136.8,132.4,112.3,110.4,108.8,69.3(t,j=27.1hz),55.7,49.9,47.5,26.4,25.4.
[0155]
hrms(esi)m/z calculated for c
13h14
cl2dnnao2[m+na
+
]311.0435,found 311.0432.
[0156]
将0.2mmol所得的化合物i、0.8mmol四氟硼酸银,100mg硅藻土,100微升水,1毫升甲苯加入10毫升的反应管中,充入氩气,密封,在100摄氏度剧烈搅拌。通过薄层色谱法监测反应,反应结束后冷至室温,混合物直接进行柱层析,分离得到白色固体(ii),收率为67%。
[0157]
化合物(ii)为:
[0158]1h nmr(400mhz,cdcl3)δ6.81
–
6.76(m,3h),3.78(s,3h),3.24(s,3h),2.97(d,j=17.2hz,1h),2.90(d,j=17.2hz,1h),1.41(s,3h).
[0159]
13
c nmr(101mhz,cdcl3)δ198.3(t,j=26.8hz),178.9,155.8,136.4,133.9,111.9,110.1,108.4,55.5,50.1(t,j=3.6hz),45.1,26.2,23.7.
[0160]
hrms(esi)m/z calculated for c
13h14
dnnao3[m+na
+
]257.1007,found 257.1006.
[0161]
将0.8mmol所得的化合物ii,8mmol甲氨盐酸盐,8mmol三乙胺,730mg无水硫酸镁,45ml的干燥甲苯加入100ml的瓶子中,用氩气吹扫三分钟除去空气。瓶子密封,在室温下反应16小时。随后加入8mmol的四氢铝锂,在80摄氏度回流反应1.5小时。反应结束后冷至室温,用乙酸乙酯萃取,对所得的有机相浓缩和柱层析得无色油状物(i-13,氘代(
±
)-esermethole,一对非对映异构体),收率为83%。
[0162]
化合物(i-13)为:
[0163]1h nmr(400mhz,cdcl3)δ6.66
–
6.63(m,2h),6.35(d,j=8.1hz,1h),4.06(s,0.52h),4.05(s,0.43h),3.74(s,3h),2.89(s,3h),2.70(t,j=5.2hz,0.51h),2.61(t,j=7.5hz,0.58h),2.53(s,3h),1.94(d,j=7.1hz,2h),1.43(s,3h).
[0164]
13
c nmr(101mhz,cdcl3;values in brackets were corresponding to the same carbon in diastereoisomers)δ152.9,146.5(146.4),138.2,112.1,109.7,107.4,98.3(98.2),55.9,52.8(td,j=21.4,3.1hz),52.7,40.7(40.6),38.1(38.0),37.9(37.8),27.4(27.3).
[0165]
hrms(esi)m/z calculated for c
14h20
dn2o[m+h
+
]234.1711,found 234.1711.
[0166]
实施例14:
[0167]
氘代(
±
)-physostigmine(i-14)的制备
[0168]
将0.5mmol所得的i-13溶解于10ml超干二氯甲烷中,在0摄氏度缓慢滴加1.5ml 1m三溴化硼的二氯甲烷溶液,在室温反应1.5小时。反应结束后冷至0摄氏度,加10ml水淬灭。用二氯甲烷萃取,所得有机相经干燥和浓缩得到粗产物酚,该粗产物酚可直接用于下一步。
[0169]
将所得的粗产物酚溶解于10ml干燥四氢呋喃中,在0摄氏度缓慢加入44mg氢化钠(60wt%),10分钟后加入95mg n-succinimidyl-n-methylcarbamate,并在室温反应2小时。反应结束用水淬灭,用乙酸乙酯萃取。有机相经干燥、浓缩和柱层析得到浅黄色油状物(i-14,氘代(
±
)-physostigmine,一对非对映异构体),两步收率为67%。
[0170]
化合物(i-14)为:
[0171]1h nmr(400mhz,cdcl3)δ6.79(dd,j=8.4,2.2hz,1h),6.75(d,j=2.2hz,1h),6.33(d,j=8.4hz,1h),4.97(d,j=3.4hz,1h),4.16(s,0.50h),4.15(s,0.49h),2.91(s,3h),2.86(s,1.29h),2.85(s,1.50h),2.73(t,j=4.9hz,0.65h),2.61(t,j=7.6hz,0.63h),2.53(s,3h),1.95(d,j=7.2hz,2h),1.42(s,3h).
[0172]
13
c nmr(101mhz,cdcl3;values in brackets were corresponding to the same carbon in diastereoisomers)δ156.1,149.4(149.3),143.2,137.3,120.5,116.1,106.6,97.8(97.7),52.7(t,j=20.5hz),52.6,40.5(40.4),38.0(37.9),37.1(37.0),27.7,27.2(27.1).
[0173]
hrms(esi)m/z calculated for c
15h21
dn3o2[m+h
+
]277.1769,found 277.1767.
[0174]
实施例15:
[0175]
氘代(
±
)-lansai b(i-15)的制备
[0176]
将1mmol i-9、5mmol四氟硼酸银、500mg硅藻土、500微升水和5ml甲苯加入反应管中,用氩气吹扫三分钟,密封,置于100摄氏度油浴锅中剧烈搅拌2小时。反应结束,冷至室温,直接进行柱层析得到无色油状物iii,收率为53%。
[0177]
化合物(iii)为:
[0178]1h nmr(400mhz,cdcl3)δ7.29
–
7.25(m,1h),7.18(dd,j=7.3,0.7hz,1h),7.04(td,j=7.6,0.8hz,1h),6.88(d,j=7.8hz,1h),3.25(s,3h),2.96(d,j=1.7hz,2h),1.41(s,3h).
[0179]
13
c nmr(101mhz,cdcl3)δ198.4(t,j=26.9hz),179.3,143.0,132.6,128.1,122.5,122.2,108.2,50.2(t,j=3.7hz),44.7,26.2,23.8.
[0180]
hrms(esi)m/z calculated for c
12h12
dnnao2[m+na
+
]227.0901,found 227.0903.
[0181]
将7.5mmol化合物iii、0.38mmol吡啶对甲苯磺酸盐、37.5mmol无水硫酸镁、15mmol叔丁基亚磺酰胺溶于20ml干燥二氯甲烷中,密封,室温搅拌过夜。反应结束,过滤除去无机盐,经短硅胶柱过滤除去吡啶对甲苯磺酸盐和叔丁基亚磺酰胺。滤液浓缩,直接用于下一步。
[0182]
所得的亚胺溶于15ml干燥四氢呋喃中,冷至零下65摄氏度,缓慢滴加1m的乙烯基氯化镁的四氢呋喃溶液,反应2小时,反应结束加入饱和氯化铵溶液淬灭,用乙酸乙酯萃取,有机相合并、干燥、浓缩,直接用于下一步。
[0183]
将所得的亚磺酰胺溶于20ml甲醇中,加入3.8ml的4m盐酸水溶液,在室温反应,薄层色谱法监测。反应结束,浓缩除去溶剂,降入碳酸氢钠水溶液碱化,使用二氯甲烷萃取。向所得的有机相中加入1m稀盐酸溶液,反复萃取直至有机相中监测不到胺。将水相碱化,使用二氯甲烷萃取,干燥,浓缩,直接用于下一步。
[0184]
将所得的胺溶于20ml干燥四氢呋喃中,冷至0摄氏度,滴加22.5ml的1m二异丁基氢化铝溶液,在0摄氏度反应1小时,随后升至室温反应2小时,随后在80摄氏度反应15小时。反应结束后冷至室温,加入饱和酒石酸钠钾水溶液淬灭,使用乙酸乙酯进行萃取,有机相合并、干燥、浓缩,直接用于下一步。
[0185]
将所得的环化产物溶解于10ml干燥四氢呋喃中,加入2ml boc酸酐,室温过夜反。反应结束,浓缩,柱层析,得无色油状物(iv,一对非对映异构体),五步收率为13%。
[0186]
化合物(iv)为:
[0187]1h nmr(400mhz,cdcl3;compound exists as a mixture of diastereomers,the major is denoted by*,minor denoted by
§
)δ7.13
–
7.09(m,1h*
§
),6.70
–
6.98(m,1h*
§
),6.69(t,j=7.2hz,0.87h*),6.66
–
6.64(m,0.15h
§
),6.43(d,j=7.8hz,0.82h*),6.38(d,j
=7.8hz,0.18h
§
),5.88(dd,j=15.8,10.5hz,0.78h*),5.48(dd,j=17.0,10.0hz,0.26h
§
),5.23(s,1h*
§
),5.07
–
5.01(m,1.91h*
§
),4.84(d,j=10.0hz,0.20h
§
),3.01(s,2.55h*),2.95(s,0.62h
§
),2.30(d,j=12.7hz,0.90h*),2.18(d,j=12.8hz,0.22h
§
),2.01(d,j=12.8hz,0.25h
§
),1.85(d,j=12.8hz,0.92h*),1.53(s,8.84h*
§
),1.45(s,2.72h*
§
),1.39(s,0.55h*
§
).
[0188]
13
c nmr(101mhz,cdcl3;compound exists as a mixture of diastereomers,the major is denoted by*,minor denoted by
§
)δ155.2
§
,150.0*,149.6
§
,146.7*,140.8*,139.3
§
,135.0*,134.9
§
,128.1*,127.8
§
,121.6
§
,121.4*,117.7*,117.3
§
,114.3
§
,113.4*,106.7*,106.1
§
,90.7*,90.3
§
,85.0*,80.0
§
,60.7*(t,j=22.0hz),50.0*,46.3*,45.1
§
,35.2*,32.7
§
,28.2
§
,27.3*,24.5*,24.1
§
.
[0189]
hrms(esi)m/z calculated for c
19h26
dn2o2[m+h
+
]316.2130,found 316.2128.
[0190]
将1.9mmol所得的化合物iv、0.38mmol二水锇酸钾、3.8mmol n-甲基吗啉-n-氧化物溶于15ml四氢呋喃和5ml水中。密封,在室温反应15小时,加入3.8mmol的高碘酸钠,反应2小时。反应结束,加入10ml硫代硫酸钠的水溶液,用乙酸乙酯萃取,合并的有机相经干燥、浓缩、柱层析得到无色油状物(v,一对非对映异构体,每个非对映异构体又包含两个旋转异构体),收率为52%。
[0191]
化合物(v)为:
[0192]1h nmr(400mhz,cdcl3;compound exists as a mixture of diastereomers,the major is denoted by*,minor denoted by
§
)δ9.54(s,0.26h*),9.51(s,0.50h*),9.22(s,0.14h
§
),7.16
–
7.10(m,1h*
§
),7.01
–
6.94(m,1h*
§
),6.73
–
6.67(m,1h*
§
),6.46
–
6.43(m,1h*
§
),5.30(s,0.51h*),5.27(s,0.11h
§
),5.14(s,0.06h
§
),5.11(s,0.27h*),3.05(s,1.51h*),3.02(s,0.12h
§
),3.01(s,0.29h
§
),2.98(s,0.89h*),2.36(d,j=13.2hz,0.58h*),2.31
–
2.25(m,0.44h*
§
),2.18(d,j=12.8hz,0.19h
§
),2.03(d,j=13.5hz,0.59h*),1.98(d,j=13.3hz,0.31h*),1.55(s,3h*
§
),1.45(s,5.45h*
§
),1.42(s,1.59h*
§
),1.40(s,1.81h*
§
).
[0193]
13
c nmr(101mhz,cdcl3;compound exists as a mixture of diastereomers,the major is denoted by*,minor denoted by
§
)δ200.8
§
,199.3*,198.9*,155.0
§
,154.4*,154.2*,149.8*,149.7*,133.7*,133.6*,133.2
§
,128.8*,128.6*,128.4
§
,122.2
§
,122.0
§
,121.7*,121.6*,118.2*,118.1*,107.1*,107.0*,90.8*,90.5
§
,90.3*,90.0
§
,81.7*,81.6*,81.4
§
,81.3
§
,65.4*(t,j=23.0hz),65.2*(t,j=23.0hz),52.1*,51.0
§
,50.6*,41.0*,40.7
§
,40.1*,35.1*,34.7*,33.5
§
,29.7
§
,28.3*,28.2
§
,28.1*,24.4*,24.1*,23.0
§
.
[0194]
hrms(esi)m/z calculated for c
18h23
dn2nao3[m+na
+
]340.1742,found 340.1741.
[0195]
将0.41mmol所得化合物v溶解于四氢呋喃/水/叔丁醇(体积比为5:5:1.3)的混合溶液中,加入2.5ml 2-甲基-2-丁烯、0.82mmol亚氯酸钠(80wt%)、2.05mmol磷酸二氢钾,在室温搅拌3小时。反应结束,加入10ml饱和氯化铵溶液,用乙酸乙酯萃取,有机相合并、干燥、
浓缩,直接用于下一步。
[0196]
将所得的羧酸溶于5ml干燥乙腈中,加入0.82mmol三甲基碘硅烷,在室温反应1小时;再加入0.82mmol三甲基碘硅烷,反应1小时。反应结束后,加入2ml饱和硫代硫酸钠溶液和10ml水,使用乙酸乙酯萃取,确认产物在水相中。舍弃有机相,将1m盐酸水溶液缓慢滴加到水相中,调节水相ph约为6,将水相浓缩,在油泵上过夜。随后加入二氯甲烷,搅拌形成悬浊液,过滤得到滤液,浓缩,干燥,直接用于下一步。
[0197]
将所得的氨基酸与0.21mmol预先制备的另一氨基酸片段(根据已有文献合成,angew.chem.int.ed.2014,53,6206)溶于6ml干燥二氯甲烷中,在0摄氏度下加入0.97mmol异丙基二乙基胺、0.84mmol双(2-氧代-3-恶唑烷基)次磷酰氯,在室温反应13小时。反应结束,加入饱和碳酸钠溶液淬灭,使用乙酸乙酯萃取,将有机相合并、干燥、浓缩、柱层析,得浅黄色固体(i-15,氘代(
±
)-lansai b),三步收率为13%。
[0198]
化合物(i-15)为:
[0199]1h nmr(400mhz,cdcl3)δ7.11
–
7.06(m,2h),7.04(d,j=7.0hz,1h),7.01(d,j=1.8hz,1h),6.70(t,j=7.4hz,1h),6.34(d,j=7.8hz,1h),6.28(d,j=8.2hz,1h),5.97(dd,j=17.4,10.6hz,1h),5.44(s,1h),5.42(s,1h),5.02(dd,j=11.9,1.3hz,1h),4.98(dd,j=5.1,1.3hz,1h),4.17(dd,j=11.2,6.1hz,1h),2.98(s,3h),2.95(s,3h),2.73
–
2.68(m,2h),2.19
–
2.13(m,2h),1.47(s,3h),1.46(s,3h),1.34(s,6h).
[0200]
13
c nmr(101mhz,cdcl3)δ165.8,165.5,150.1,148.6,148.2,138.7,132.9,132.8,128.7,126.3,122.3,120.2,118.1,110.1,105.8,105.4,86.9,86.5,60.1,50.5,50.3,42.7,42.6,40.7,33.1,32.9,28.5,25.5,25.4.
[0201]
13
c nmr(176mhz,cdcl3)δ165.8,165.7,149.9,148.4,147.5,139.6,133.2,133.0,128.8,126.5,122.4,120.3,118.3,110.3,106.3,106.1,87.1,86.7,60.2,59.8(t,j=21.9hz),50.5,50.3,42.7,42.6,40.7,33.8,33.1,28.5,25.6,25.5.
[0202]
hrms(esi)m/z calculated for c
31h35
dn4nao2[m+na
+
]520.2793,found 520.2793.
[0203]
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1