一种用于电催化合成氨的碳化钼-碳化硼复合材料的制备方法及其产品和应用
1.本发明涉及电催化氮还原电极材料的领域,特别涉及一种用于电催化合成氨的碳化钼-碳化硼复合材料的制备方法及其产品和应用。
背景技术:
2.氨(nh3)是工业上使用最广泛的无机化学品之一,被广泛应用于肥料生产、药物合成及许多其他工业领域。此外,氨具有高能量密度(5.52kw h kg-1
)和17.6wt%的氢,因此氨具有作为储氢载体及“无碳燃料”的应用前景。当前,全世界的工业合成氨几乎完全依赖于传统的haber-bosch工艺,该工艺在高温(400~600℃)高压(20~30mpa)下进行,每年消耗全球约2%的化石能源,排放的co2约占全球总排放量的1.4%,对环境造成极大影响。因此,开发环境友好型的nh3合成方法势在必行。电化学合成氨(e-nrr)利用可再生能源(如太阳能或风能)产生的电力,在环境条件下通过非均相催化剂将n2电化学还原为nh3。从热力学上讲,温和清洁的e-nrr比haber-bosch过程的能效高20%。还可以实现氨的分散生产以降低运输成本,因此是传统合成氨工艺最有希望的替代方法之一。其中,设计高性能e-nrr催化剂强化n2的吸附和活化至关重要。
3.文献“a theoretical evaluation of possible transition metal electro-catalysts for n
2 reduction”通过理论计算表明金属mo是位于e-nrr活性火山图顶峰的元素。
4.公开号为cn113058658a的中国发明专利公开了一种超疏水负载钼催化剂的制备方法,利用具有丰富氧配位点的磷钨酸为载体,以五水氯化钼为修饰物种的前驱物,开发出高性能的电催化固氮用超疏水负载钼物种的磷钨酸电催化剂。
5.公开号为cn113215598a的中国发明专利公开了一种用于电催化合成氨的bi-mos2纳米复合材料,以二硫化钼为基底,通过水热反应将铋纳米晶体与二硫化钼纳米片进行复合。用于电催化合成氨。
6.然而mo基材料(如mo2c)同时也表现出优异的her催化性能,这限制了钼基催化剂在e-nrr催化领域的应用。因此,如何提高其催化性能是目前本领域亟需解决的技术问题。
技术实现要素:
7.本发明的目的在于提供一种用于电催化合成氨的碳化钼-碳化硼复合材料的制备方法及其产品和应用,采用该制备方法制备得到的mo2c-b4c复合材料具有优于碳化钼和碳化硼的界面结构及电化学性能,应用在电催化合成氨上能够有效提高电催化合成氨产率和法拉第效率。
8.本发明提供如下技术方案:
9.一种用于电催化合成氨的碳化钼-碳化硼复合材料的制备方法,所述制备方法为:将mo2c和b4c球磨处理后进行煅烧,得到mo2c-b4c复合材料。
10.所述mo2c和b4c中mo与b的摩尔比为1:1~1:4。
11.mo2c和b4c中mo与b的摩尔比会影响催化剂中mo-b键数量,mo-b键对催化性能具有积极作用。优选地,所述mo2c和b4c中mo与b的摩尔比为1:2,这时形成最多的mo-b键。
12.所述球磨的时间为1~16h,球料比为20:1~30:1,转速为20-60hz。球磨促进两相混合,同时改变颗粒粒径,球磨时间过短,颗粒较大;球磨时间过长,颗粒不再进一步减小,并可能造成催化剂结构改变,不利于电催化。
13.优选地,球磨时间为3~6小时。mo2c-b4c复合材料作为催化剂具有较大的电化学活性表面积及较高的电导率。进一步优选地,球磨时间为4小时,在球磨4h条件下催化剂具有最大的电化学活性表面积及最高电导率,因而活性最佳。
14.所述煅烧在非氧气氛下进行,比如氮气气氛下。煅烧的温度为450~750℃,升温速率为2-10℃/min,时间为1~10h。本发明通过煅烧促进mo2c和b4c两相化学键合,使煅烧后的复合材料具有更高的电化学活性表面积及高电导率,有利于电催化合成氨。优选地,所述煅烧的气氛为n2,所述煅烧的温度为450~550℃,升温速率为5℃/min,时间为4h。
15.其中,所述制备方法还包括对煅烧后的产物进行洗涤干燥:置于0.5mol/l稀硫酸中,超声1h,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。将催化剂置于真空干燥箱中干燥,于40℃下干燥10小时,随后研磨至粉末,装入样品瓶中保存。
16.本发明还提供了一种根据上述制备方法得到的碳化钼-碳化硼复合材料。
17.所述碳化钼-碳化硼复合材料中b4c部分包覆于mo2c表面,对mo2c表面进行修饰,两相间生成mo-b键。
18.在本发明提供的碳化钼-碳化硼复合材料中,b4c部分包覆于mo2c表面,具有丰富的缺陷位点,并且能够吸附大量n2,两相间大量mo-b键的形成有效促进了电化学过程的发生。
19.本发明还提供了一种上述碳化钼-碳化硼复合材料在电催化合成氨上的应用。
20.富含缺陷的mo2c-b4c复合材料暴露的b-mo双位点与单一位点相比有更强n2吸附作用和更强的电荷转移能力。因此,钼、硼的协同作用使复合材料具有优异的电催化合成氨性能。
21.与现有技术相比,本发明通过采用球磨、煅烧方法合成碳化钼-碳化硼复合材料,通过工艺过程控制,使其具有优于碳化钼和碳化硼的界面结构及电化学性能,能够有效提高电催化合成氨产率和法拉第效率,同时在5次循环测试中具有良好的稳定性。
22.本发明提供的mo2c-b4c复合材料将硼与钼基催化剂进行复合,减弱mo表面的析氢能力,同时利用钼基与硼基材料的协同催化作用,实现有效的e-nrr催化。
23.本发明提供的mo2c-b4c复合材料氨产率可达~8μg h-1
mg-1cat.
,法拉第效率可达~18%(ph=1的0.1m hcl电解液)。
附图说明
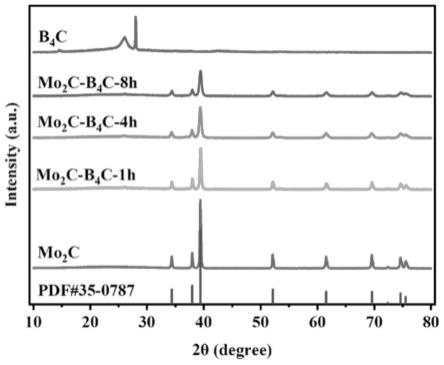
24.图1为实施例1中所制备的mo2c-b4c复合材料的x射线衍射谱图;
25.图2为实施例1中所制备的mo2c-b4c复合材料的透射电镜图;
26.图3为实施例1中所制备的mo2c-b4c复合材料的x射线光电子能谱图;
27.图4为实施例1中所制备的mo2c-b4c复合材料的氮气吸脱附曲线;
28.图5为实施例1中所制备的mo2c-b4c复合材料及mo2c和b4c用于电催化合成氨的氨产率;
29.图6为实施例1中所制备的mo2c-b4c复合材料的氨产率和法拉第效率图;
30.图7为实施例1-3中所制备的mo2c-b4c复合材料用于电催化氮还原的nh3产率对比图;
31.图8为实施例1、4、5、6中所制备的mo2c-b4c复合材料用于电催化氮还原的nh3产率对比图;
32.图9为实施例1、7、8、9中所制备的mo2c-b4c复合材料用于电催化氮还原的nh3产率对比图;
33.图10为实施例1、4、5、6所制备的mo2c-b4c复合材料的电化学活性表面积;
34.图11为实施例1、4、5、6所制备的mo2c-b4c复合材料的电化学阻抗;
35.图12为实施例1、7、8、9所制备的mo2c-b4c复合材料的电化学活性表面积;
36.图13为实施例1、7、8、9所制备的mo2c-b4c复合材料的电化学阻抗;
37.图14为实施例1-3所制备的mo2c-b4c复合材料经xps测试得到的硼的不同成键类型及b-mo键占比。
具体实施方式
38.下面对本发明的具体实施方式进行详细说明,但是需要指出的是,本发明的保护范围并不受这些具体实施方式的限制,而是由附录的权利要求书来确定。以下实施例和对比例中,如无特别说明,所用的原料均可通过商购获得。
39.本说明书提到的所有出版物、专利申请、专利和其它参考文献全都引于此供参考。除非另有定义,本说明书所用的所有技术和科学术语都具有本领域技术人员常规理解的含义。在有冲突的情况下,以本说明书的定义为准。
40.以下采用实施例进一步详细地说明本发明,但本发明并不限于这些实施例。
41.本发明的mo2c-b4c复合材料具有优异的电催化合成氨性能,将5mg催化剂样品分散到900μl乙醇中,加入100μl nafion溶液形成均匀得浆液,超声1h,然后将40μl混合溶液滴涂到预处理的碳纸中,负载浓度为0.2mg cm-2
,作为工作电极,可进行电催化合成氨测试。
42.本发明的i-t测试,是使用chi660e电化学工作站,在ph=1的0.1m hcl电解液中进行的,以ag/agcl(3m kcl)作为参比电极,碳棒为对电极,上述催化剂制备的电极为工作电极。
43.在本说明书的上下文中,包括在以下的实施例和对比例中,电催化合成氨反应的催化性能通过产率和法拉第效率和稳定性来考察。
44.氨产率=测得的电解液中氨浓度
×
电解液的体积/催化剂的质量/反应时间。
45.法拉第效率=测得的电解液中氨浓度
×
电解液的体积
×
96485
×
3/电荷量
×
100%。
46.实施例1
47.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨4小时。
48.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速
率升温至550℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
49.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
50.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
51.实施例2
52.(1)将0.816g碳化钼和0.110g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨4小时。
53.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至550℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
54.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
55.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
56.实施例3
57.(1)将0.816g碳化钼和0.442g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨4小时。
58.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至550℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
59.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
60.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
61.实施例4
62.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨1小时。
63.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至550℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
64.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
65.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
66.实施例5
67.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨8小时。
68.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至550℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
69.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
70.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
71.实施例6
72.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨16小时。
73.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至550℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
74.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
75.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
76.实施例7
77.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨4小时。
78.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至450℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
79.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
80.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
81.实施例8
82.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨4小时。
83.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至650℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
84.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
85.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
86.实施例9
87.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨4小时。
88.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至750℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
89.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
90.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
91.对比例1
92.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于研钵中手动研磨30分钟。
93.(2)将(1)得到的黑色粉末置于瓷舟中进行煅烧,在n2气氛下以5℃/min的升温速率升温至550℃并维持4小时,待管式炉冷却至室温后取出瓷舟,得到黑色固体粉末。
94.(3)将(2)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
95.(4)将(3)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
96.对比例2
97.(1)将0.816g碳化钼和0.221g纳米碳化硼加入球磨罐中,于行星式球磨机中在40hz转速下球磨4小时。
98.(2)将(1)得到的黑色粉末置于稀硫酸中,超声1小时,抽滤洗涤至中性,以充分洗净未反应的物质并尽可能排除掉催化剂中的氨污染。
99.(3)将(2)得到的固体粉末置于真空干燥箱中,于40℃下干燥10小时,研磨至粉末,得到mo2c-b4c。
100.对比例3
101.商业碳化钼。
102.对比例4
103.商业纳米碳化硼。
104.试验例1
105.xrd测试:将实施例1制备得到的mo2c-b4c复合材料样品进行xrd测试,如图1所示。x射线衍射峰能够和mo2c的标准卡片对应,同时显示出与b4c一致的弱衍射峰,表明mo2c-b4c复合材料的成功合成,且其中b4c相的结晶度较低。
106.透射电子显微镜:将实施例1得到的mo2c-b4c复合材料进行透射电子显微镜测试,如图2所示。高分辨率tem图像显示出mo2c与b4c紧密结合,证明依据上述实验方法合成的mo2c-b4c复合材料中mo2c与b4c成功复合。
107.试验例2
108.xps测试:将实施例1制备得到的mo2c-b4c复合材料样品进行xps测试,如图3所示。与b4c和mo2c-b4c(未煅烧)相比,mo2c-b4c(煅烧后)其b1s谱图低结合能处的峰明显向左偏移,表明煅烧过程中有大量b-mo键形成。b-mo键的形成代表b4c与mo2c之间不是简单地物理混合,两相界面形成配位键,有利于界面间电子转移。
109.n
2-tpd测试:测试实施例1和对比例2-4样品的氮气吸脱附曲线,如图4所示。b4c和mo2c-b4c都表现出一定的氮吸附能力,煅烧后的mo2c-b4c的解吸峰位于415℃,明显高于煅烧前365℃,同时,煅烧后的mo2c-b4c拥有更大的解吸峰面积。煅烧后mo2c-b4c复合材料能够更紧密地吸附大量n2,说明该材料暴露b-mo双位点与单一位点相比有更强n2吸附作用。
110.试验例3
111.电化学性能测试:将实施例1制得的mo2c-b4c复合材料组装成电极在三电极体系下进行电化学性能测试。
112.将5mg催化剂样品分散到900μl乙醇中,加入100μl nafion溶液形成均匀得浆液,
超声1h,然后将40μl混合溶液滴涂到预处理的碳纸中,负载浓度为0.2mg cm-2
,作为工作电极;以ag/agcl(3m kcl)作为参比电极,碳棒为对电极。使用chi660e电化学工作站,在ph=1的0.1m hcl电解液中进行。
113.图5为测试实施例1和对比例3、4用于电催化氮还原的nh3产率。实施例1具有远高于对比例3/4的nh3产率,证明钼、硼的协同催化作用。
114.图6为实施例1制得的mo2c-b4c复合材料在不同电势下的nh3产率和法拉第效率图,其中曲线为合成氨的法拉第效率,柱状图为nh3产率。由图6可知,本发明的mo2c-b4c复合材料具有良好的电催化合成氨的活性和优越的选择性。
115.图7为实施例1-3用于电催化氮还原的nh3产率,考察mo和b的比例对电催化氮还原活性的影响。当mo:b=1:1时,过少的b4c导致mo2c表面氢吸附位点过多暴露,体系中发生过多析氢竞争反应因而降低了电催化氮还原的选择性。当mo:b=1:4时,过多的b4c覆盖于mo2c表面,虽然有利于抑制析氢反应,但其表面的mo与b接界的活性位点数量减少,进而使其电催化氮还原活性受到抑制。
116.图8为实施例1、4、5、6用于电催化氮还原的nh3产率。考察合成工艺中球磨时间对电化学氮还原性能的影响,其中实施例1展现出最佳的电催化氮还原活性。
117.图9为实施例1、7、8、9用于电催化氮还原的nh3产率。考察合成工艺中煅烧温度对电化学氮还原性能的影响,其中实施例1展现出最佳的电催化氮还原活性。
118.图10为实施例1、4、5、6所制备的mo2c-b4c复合材料的电化学活性表面积。实施例1显示出3.07mf cm-2
的电化学活性表面积,高于实施例4(1.75mf cm-2
)、实施例5(2.13mf cm-2
)和实施例6(2.42mf cm-2
),揭示了实施例1大量活性位点有利于提高反应活性。
119.图11为实施例1、4、5、6的电化学阻抗nyquist图,实施例1与实施例4、5、6相比具有最小的曲率半径,进一步证明了将mo2c和b4c两相充分混合并调控其相界面及颗粒尺寸有利于电化学过程中的电子转移。更快的将电子转移到吸附的n2可以加速电催化氮还原过程。
120.图12为实施例1、7、8、9的电化学活性表面积。在450℃和550℃下煅烧得到的实施例1和实施例7具有近乎相同的电化学活性表面积,随煅烧温度升高至650℃和750℃,实施例8和实施例9电化学活性表面积降低至实施例1的82%。
121.图13为实施例1、7、8、9的电化学阻抗。在450℃和550℃下煅烧得到的实施例1和实施例7具有近乎相同的电导率,随煅烧温度升高至650℃和750℃,nyquist图中曲率半径同样显著增加。因此,合成过程中过高的煅烧温度不利于催化剂的电催化氮还原性能。
122.图14为实施例1-3经xps测试得到的硼的不同成键类型及b-mo键占比,实施例1中b-mo键占比32.9%,高于实施例2的28.8%和实施例3的24.9%,由于硼和钼具有不同的电子接受和回馈能力,b-mo活性中心将极大地促进非极性n≡n键的极化和断裂,进而提高电催化氮还原催化活性。
123.以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1