一种4-溴吡唑类化合物的制备方法
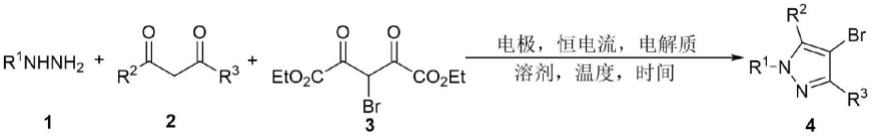
1.本发明涉及有机合成技术领域,特别的涉及电化学促进下多组分“一锅法”制备4-溴吡唑类化合物的方法。
背景技术:
2.吡唑衍生物的基本构型广泛存在于天然产物和药物分子中,对药物化学和材料科学具有重要意义。因此,它们的构建和功能化引起了合成有机化学家的极大关注。在众多吡唑衍生物中,4-溴吡唑类化合物不仅表现出广泛的生物、药物活性,也可作为合成转化的通用合成中间体。
3.目前,报道的制备4-溴吡唑类化合物的方法主要包括以溴化钠作为溴源的氧化溴化反应、以溴素作为溴源的溴化反应、光催化溴化反应和电催化溴化反应等。2017年,mackay课题组报道了以溴化钠作为溴化试剂,在过量过氧单磺酸钾(oxone)氧化剂促进下的溴化反应制备4-溴吡唑类化合物。该方法需要使用过量的氧化剂,不符合原子经济性要求。以溴素作为溴源的溴化反应也有报道,如发明专利wo2007058832公开了一种以溴素作为溴化剂的制备4-溴吡唑类化合物的方法。该方法使用腐蚀性强、毒性大的溴素作为溴化试剂,不适用于工业化生产。2018年,koenig课题组报道了以溴化钠作为溴源,可见光促进下吡唑发生溴化反应制备4-溴吡唑类化合物的方法。但该方法需要使用特殊的光催化剂,成本较高。2019年,雷爱文课题组报道了一种电化学促进下吡唑衍生物溴化反应制备4-溴吡唑类化合物的方法。该方法以廉价的溴化钠作为溴源,以电作为清洁能源,在温和的反应条件下便可以实现吡唑衍生物的溴化反应。但由于溴离子的氧化态较低,在阳极表面容易发生过氧化效应,因此该反应需要使用2-4当量的溴化剂才能使反应有效进行。
4.综上所述,已报道的制备4-溴吡唑类化合物的方法均以吡唑作为起始原料,多组分“一锅法”环化溴化方法尚未报道。且上述反应均存在一定的局限性,如所用试剂毒性大、环境污染严重、后处理复杂、试剂价格昂贵、溴化率低等。
技术实现要素:
5.针对上述现有技术的不足,本发明的目的在于提供一种4-溴吡唑类化合物的制备方法,解决现有方法需要使用大量氧化剂、有毒有害试剂或特殊金属催化剂以及操作复杂、环境污染严重等问题。
6.为了解决上述技术问题,本发明采用了如下的技术方案:
7.一种4-溴吡唑类化合物的制备方法,其特征在于,包括以下步骤:将肼类化合物(1)、1,3-丙二酮类化合物(2)、2-溴丙二酸二乙酯(3)和电解质置于有机溶剂中得到反应液,然后将两个电极置于反应液中进行恒电流电解,充分反应后,经后处理得到4-溴吡唑类化合物(4);
[0008][0009]
其中r1为h、烷基、芳基或芳香杂环;r2、r3均为烷基或芳基。
[0010]
进一步,所述r1为c
1-c3的烷基、c
1-c3的烷氧基、卤素或卤素取代的c
1-c3烷基。
[0011]
进一步,所述r1为c
1-c3的烷基、芳基、c
1-c3烷基取代的芳基或卤素取代的芳基;
[0012]
r2为卤素、c
1-c3的烷基、卤素取代的c
1-c3烷基或c
1-c3的烷氧基;
[0013]
r3为卤素、c
1-c3的烷基、卤素取代c
1-c3的烷基或c
1-c3的烷氧基。
[0014]
进一步,所述肼类化合物、1,3-丙二酮类化合物、2-溴丙二酸二乙酯的摩尔比为:1-3:1:1-3。
[0015]
进一步,所述后处理包括以下步骤:反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离得到4-溴吡唑类化合物;所述柱层析中洗脱溶剂为乙酸乙酯/石油醚。
[0016]
进一步,所述反应温度为25~80℃;反应时间为10~24h。
[0017]
进一步,所述恒电流强度为2~30ma。
[0018]
进一步,所述电极为铂片、石墨片、铜片、不锈钢、镍片、铝片、泡沫镍或泡沫铝中的一种或两种。
[0019]
进一步,所述电解质为四丁基四氟硼酸铵、四乙基四氟硼酸铵、四氟磷酸钾、高氯酸锂、碘化钾、溴化铵、四丁基碘化铵、四乙基六氟磷酸铵、四乙基高氯酸铵、六氟磷酸钠中的一种或多种。
[0020]
进一步,所述有机溶剂为乙腈、丙酮、乙醇、n,n-二甲基甲酰胺、二甲基亚砜、二氯乙烷、二氯甲烷、甲苯中的一种或多种。
[0021]
其中,肼类化合物和1,3-丙二酮类化合物来源广泛,价格低廉,因此选取作为其实原料。
[0022]
相比现有技术,本发明具有如下有益效果:
[0023]
1、本发明制备方法以肼、1,3-丙二酮和2-溴丙二酸二乙酯作为原料,在电解条件下,首先经环化反应得到吡唑化合物,后经电催化溴化生成4-溴吡唑类化合物;该反应为多组分“一锅法”反应,一步反应同时形成三个新化学键,较分步反应相比,多组分反应具有更高的效率,且避免了复杂的后处理过程。
[0024]
2、本发明4-溴吡唑类化合物制备方法,以电子作为清洁试剂,避免了大量氧化剂的使用;以2-溴丙二酸二乙酯作为溴化试剂,与溴化钠作为溴源的方法相比,2-溴丙二酸二乙酯具有较高的氧化态,难以被阳极氧化,从而避免了过氧化效应而降低了溴化试剂的用量,仅使用一当量的溴化试剂便可高效获得4-溴吡唑类化合物。且本发明无需使用过渡金属催化剂和配体或大量的氧化还原剂,具有反应条件温和、操作简便、原料廉价和适用范围广泛等优点,具有优良的应用前景,为4-溴吡唑类化合物的制备提供了一种绿色的新方法。
具体实施方式
[0025]
下面结合实施例对本发明作进一步的详细说明。下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和原料,如无特殊说明,均可以从商业途径获得和/或
根据已知的方法制备获得。
[0026]
实施例1
[0027][0028]
将苯肼(1a,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4a(收率:90%)。
[0029]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.43(t,j=7.5hz,2h),7.35(d,j=7.5hz,4h),2.27(s,7h).
13
cnmr(126mhz,cdcl3)δ147.53,137.46,129.13,127.77,124.66,96.35,12.32,11.72。由此可知,制得的产物结构为目标产物4a。
[0030]
实施例2
[0031]
用pt(+)|cu(-)电极对代替pt(+)|pt(-)电极对,其余条件同实施例1,得到目标产物4a的收率为84%。
[0032]
实施例3
[0033]
用pt(+)|ni(-)电极对代替pt(+)|pt(-)电极对,其余条件同实施例1,得到目标产物4a的收率为88%。
[0034]
实施例4
[0035]
用pt(+)|al(-)电极对代替pt(+)|pt(-)电极对,其余条件同实施例1,得到目标产物4a的收率为83%。
[0036]
实施例5
[0037]
用四乙基四氟硼酸铵代替四丁基四氟硼酸铵,其余条件同实施例1,得到目标产物4a的收率为85%。
[0038]
实施例6
[0039]
用四氟磷酸钾代替四丁基四氟硼酸铵,其余条件同实施例1,得到目标产物4a的收率为80%。
[0040]
实施例7
[0041]
用高氯酸锂代替四丁基四氟硼酸铵,其余条件同实施例1,得到目标产物4a的收率为78%。
[0042]
实施例8
[0043]
用四丁基碘化铵代替四丁基四氟硼酸铵,其余条件同实施例1,得到目标产物4a的收率为62%。
[0044]
实施例9
[0045]
用四乙基高氯酸铵代替四丁基四氟硼酸铵,其余条件同实施例1,得到目标产物4a的收率为75%。
[0046]
实施例10
[0047]
用丙酮代替乙腈,其余条件同实施例1,得到目标产物4a的收率为82%。
[0048]
实施例11
[0049]
用乙醇代替乙腈,其余条件同实施例1,得到目标产物4a的收率为65%。
[0050]
实施例12
[0051]
用二甲基亚砜代替乙腈,其余条件同实施例1,得到目标产物4a的收率为90%。
[0052]
实施例13
[0053]
用n,n-二甲基甲酰胺代替乙腈,其余条件同实施例1,得到目标产物4a的收率为79%。
[0054]
实施例14
[0055]
电流强度以5ma代替10ma,其余条件同实施例1,得到目标产物4a的收率为80%。
[0056]
实施例15
[0057]
电流强度以12ma代替10ma,其余条件同实施例1,得到目标产物4a的收率为82%。
[0058]
实施例16
[0059]
电流强度以20ma代替10ma,其余条件同实施例1,得到目标产物4a的收率为78%。
[0060]
实施例17
[0061]
反应温度50℃代替室温,其余条件同实施例1,得到目标产物4a的收率为84%。
[0062]
实施例18
[0063]
反应时间延长至24h,其余条件同实施例1,得到目标产物4a的收率为88%。
[0064]
由上述实施例1-18可以看出,最佳的反应条件为实施例1的反应条件,即pt(+)|pt(-)电极作为电极对,电流强度为10ma,电解质为四丁基四氟硼酸铵,溶剂为乙腈,反应温度为室温,反应时间为12h。在最优化反应条件下,发明人进一步选取不同取代基的肼和1,3-丙二酮作为原料以发展高效的制备4-溴吡唑类化合物方法。
[0065]
实施例19
[0066][0067]
将苯肼(1a,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4a(收率:90%)。
[0068]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.43(t,j=7.5hz,2h),7.35(d,j=7.5hz,4h),2.27(s,7h).
13
cnmr(126mhz,cdcl3)δ147.53,137.46,129.13,127.77,124.66,96.35,12.32,11.72。由此可知,制得的产物结构为目标产物4a。
[0069]
实施例20
[0070][0071]
将4-甲基苯肼(1b,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,
0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4b(收率:88%)。
[0072]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.32-7.20(m,4h),2.40-2.36(m,3h),2.30-2.24(m,6h).
13
cnmr(126mhz,cdcl3)δ147.25,137.77,137.48,137.40,129.68,124.58,96.00,21.07,12.32,11.63。由此可知,制得的产物结构为目标产物4b。
[0073]
实施例21
[0074][0075]
将4-甲氧基苯肼(1c,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4c(收率:70%)。
[0076]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.44(t,j=7.5hz,2h),7.38(d,j=7.5hz,2h),2.31(d,j=2.0hz,6h).
13
cnmr(126mhz,cdcl3)δ147.59,139.85,137.53,129.17,127.83,124.71,96.38,12.34,11.75。由此可知,制得的产物结构为目标产物4c。
[0077]
实施例22
[0078][0079]
将4-氯苯肼(1d,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4d(收率:88%)。
[0080]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.41(d,j=8.0hz,2h),7.32(d,j=8.0hz,2h),2.27(d,j=5.5hz,6h).
13
cnmr(126mhz,cdcl3)δ148.00,138.32,137.50,133.52,129.32,125.72,96.82,12.31,11.75。由此可知,制得的产物结构为目标产物4d。
[0081]
实施例23
[0082][0083]
将4-三氟甲基苯肼(1e,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4e(收率:85%)。
[0084]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.72(d,j=8.0hz,2h),7.54(d,j=
8.0hz,2h),2.34(s,3h),2.31(s,3h).
13
cnmr(126mhz,cdcl3)δ148.60,142.57,137.54,129.60,129.32,126.42,126.38,126.35,126.33,124.86,124.15,123.30,122.70,97.72,12.31,12.00。由此可知,制得的产物结构为目标产物4e。
[0085]
实施例24
[0086][0087]
将苯4-肼基嘧啶(1f,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4f(收率:60%)。
[0088]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ9.21(s,1h),8.42(d,j=2.5hz,1h),8.33(s,1h),2.63(s,3h),2.30(s,3h).
13
cnmr(126mhz,cdcl3)δ149.74,149.42,141.15,141.08,141.07,139.75,138.02,99.90,13.47,12.55。由此可知,制得的产物结构为目标产物4f。
[0089]
实施例25
[0090][0091]
将环己基肼(1g,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4g(收率:80%)。
[0092]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ3.88(d,j=6.0hz,1h),2.21(d,j=9.0hz,6h),1.88(d,j=7.5hz,6h),1.71(d,j=7.5hz,1h),1.30(m,3h).
13
cnmr(126mhz,cdcl3)δ145.28,135.80,93.60,58.50,32.63,25.74,25.15,12.32,10.21。由此可知,制得的产物结构为目标产物4g。
[0093]
实施例26
[0094][0095]
将苯肼(1a,0.5mmol)、3,5-庚二酮(2b,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在室温、10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4h(收率:68%)。
[0096]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.40-7.35(m,2h),7.30(d,j=7.5hz,
3h),2.60(d,j=7.5hz,4h),1.21(t,j=7.5hz,3h),1.03(t,j=7.5hz,3h).
13
cnmr(126mhz,cdcl3)δ152.46,142.94,129.18,128.10,125.31,94.35,20.40,18.66,12.97,12.87。由此可知,制得的产物结构为目标产物4h。
[0097]
实施例27机理控制实验
[0098][0099]
为了进一步验证溴化反应的反应机理,发明人以1-苯基-3,5-二甲基吡唑5作为反应原料,在最优化条件下,考察溴化反应的情况;继而,在最优化反应条件下,向反应体系中添加2当量自由基捕获剂(bht,2,6-二叔丁基对甲酚),考察反应体系中存在的自由基情况。
[0100]
将1-苯基-3,5-二甲基吡唑5、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在10ma恒电流下电解12h,反应结束后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4a(收率:92%)。
[0101]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.43(t,j=7.5hz,2h),7.35(d,j=7.5hz,4h),2.27(s,7h).
13
cnmr(126mhz,cdcl3)δ147.52,139.93,137.45,129.12,127.76,124.65,96.34,12.32,11.72。由此可知,制得的产物结构为目标产物4a。
[0102][0103]
将苯肼(1a,0.5mmol)、乙酰丙酮(2a,0.5mmol)、2-溴丙二酸二乙酯(3,0.5mmol)、nbu4nbf4(0.5mmol)、bht(1.0mmol)和6ml乙腈置于非分离式电解池中,均以铂电极(pt)作为阳极和阴极,在10ma恒电流下电解12h。反应结束后,将反应原液经gc-ms分析,结果表明,反应液中存在化合物6(ms:378.2)。而后,将反应液减压浓缩除去溶剂,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/石油醚)得到目标产物4a(收率:84%)。
[0104]
核磁共振结果显示1hnmr(500mhz,cdcl3)δ7.43(t,j=7.5hz,2h),7.33(d,j=7.5hz,3h),2.26(s,6h).
13
cnmr(126mhz,cdcl3)δ147.51,139.92,137.44,129.11,127.75,124.64,96.33,12.31,11.71。由此可知,制得的产物结构为目标产物4a。
[0105]
由上述实验结果可知,1-苯基-3,5-二甲基吡唑5为溴化反应过程中的重要中间体,且反应体系中存在丙二酸二乙酯自由基(其与bht自由基捕获剂结合生成化合物6)。因此,本发明可能的反应机理为:首先,苯肼(1a)和乙酰丙酮(2a)发生环化反应获得1-苯基-3,5-二甲基吡唑(5)中间体。而2-溴丙二酸二乙酯(3)经阴极还原生成自由基阴离子,继而释放出br-和丙二酸二乙酯自由基。在阳极表面,1-苯基-3,5-二甲基吡唑5经阳极氧化得到自由基阳离子,并与br-反应得到自由基中间体,后经阳极氧化、去质子获得目标产物4a。
[0106]
综上,本发明制备方法以肼、1,3-丙二酮和2-溴丙二酸二乙酯作为原料,在电解条件下,首先经环化反应得到吡唑化合物,后经电催化溴化生成4-溴吡唑类化合物;该反应为多组分“一锅法”反应,一步反应同时形成三个新化学键,较分步反应相比,多组分反应具有更高的效率,且避免了复杂的后处理过程。且以2-溴丙二酸二乙酯作为溴化试剂,与溴化钠
作为溴源的方法相比,2-溴丙二酸二乙酯具有较高的氧化态,难以被阳极氧化,从而避免了过氧化效应而降低了溴化试剂的用量,仅使用一当量的溴化试剂便可高效获得4-溴吡唑类化合物。且本发明底物适用性广泛、反应条件温和、绿色高效,有利于工业生产。
[0107]
以上所述仅为本发明的较佳实施例而已,并不以本发明为限制,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1