一种加压微提取设备和加压微提取方法及其应用与流程
本发明属于分析化学和天然产物化学领域,具体涉及一种加压微提取设备和基于该设备的加压微提取方法及其在药用植物的成分检测和质量控制方面的应用。
背景技术:
药用植物(包括中药)的活性成分都存在于植物组织中,只有将活性成分从植物组织中提取出来,才能开展化学结构鉴定、活性检测、质量控制等方面的研究。因此,提取技术是药用植物研究,尤其是成分分析和质量控制的基础和关键技术。
在中药成分分析和质量控制领域,传统的提取方法有索氏提取法、超声提取法、回流提取法、浸渍法等。虽然这些提取方式应用广泛,但是一次提取短则几十分钟,长则几个小时;提取的药材也以克为单位。上述传统的提取方式不仅耗时耗力,而且对药材以及有机溶剂的使用量都较大,尤其不适于名贵药材的分析,不利于环境保护。另外,很多活性成分的结构不稳定,长时间高温提取会造成这些成分降解或转化,影响甚至误导检测结果。在检测手段日趋成熟的情况下,提取显然是实现快速微量分析的关键。显然,传统的提取方法已无法满足以快速、微量、准确等为目标的中药质量控制和研究的要求。
近年来陆续出现了一些新的提取技术,如加压溶剂提取技术、超临界流体色谱技术等。如公开号cn104857739a(公开日2015年8月26日)、名称为“一种新型加压提取装置”的中国发明专利申请公开了一种加压提取装置,包括小型提取罐、平顶罐盖,所述罐盖由螺栓固定在小型提取罐顶端,所述罐盖与罐体口间设有密封圈,罐盖上设有压力表、安全阀、压缩空气进口及控制阀门、温度测量传感器、取样导管、取样控制阀、加液导管以及加液控制阀,罐体外侧有油浴或水浴加热装置,加热装置外设有保温层,罐底下设有电加热管、温度控制传感器及pid温度调节仪。该设 备虽然在加压提取过程中可随时进行取样,但是需要专门的加压提取设备,且不能实现微型化。
因此,有必要开发出消耗药材更小、耗时更短,特别是能够实现在线提取和检测联动的方法。
技术实现要素:
针对上述问题,本发明的一个目的在于提供一种加压微提取设备以及利用该设备的一种加压微提取和在线检测的方法。本发明所述的设备及方法基于现有的液相色谱仪即可实现,简便、快速,完成一次提取仅需毫克级别的样品,耗时几分钟;不仅大幅度减少了药材和溶剂的消耗,特别适合名贵药材的质量控制和成分研究,而且极大地降低了化学成分降解和结构变化的几率,从而使检测结果更真实。
为了实现上述发明目的,本发明采用了如下的技术方案:
一种加压微提取设备,包括提取柱和加压装置,所述提取柱和加压装置由管线连接;所述提取柱为液相色谱仪预柱套和置于其中的填充了待测的样品和惰性填料的预柱芯。
优选的,所述惰性填料选自硅藻土、正相硅胶、十八烷基键合硅胶、辛烷键合硅胶和纤维素中的一种或多种。
优选的,待测样品粉碎,粒径≤0.5mm。
优选的,所述加压装置为液相色谱仪的高压输液泵。
优选的,所述加压微提取设备还包括温度控制系统;提取时,所述提取柱置于所述温度控制系统中。
更优选的,所述温度控制系统为液相色谱仪的柱温箱。
本发明的另一个目的在于提供所述加压微提取设备在植物化学成分提取、定性检测和/或定量分析中的应用,包括待测植物样品与惰性填料填充于所述提取柱中,提取溶剂经所述加压设备进入所述提取柱中,收集流出液,即得待测植物提取液,然后进行化学成分分离、定性检测和/或定量分析。
优选的,定性检测和/或定量分析的检测器选自紫外检测器、荧光检测器、电化学检测器、示差折光检测器、二极管阵列检测器、蒸发光散射检测器和质谱检测器等本领域常规的检测器。
作为一个优选的实施方式,本发明提供一种红花的加压微提取方法, 具体操作为:
i.填装提取柱:红花药材粉碎,过60~80目筛,取1.0mg装于去除了填料的预柱芯中并用惰性材料硅藻土4.0~6.0mg进行填充,然后装入预柱套中置于液相色谱仪柱温箱中;
ii.加压微提取:柱温箱温度为55~65℃,以体积百分浓度25%的甲醇水溶液或体积百分浓度0.01%的甲酸水溶液为提取溶剂,开启液相高压泵,流速3~5ml/min,提取时间20~120秒;收集流出液。
优选的,以体积浓度25%的甲醇水溶液为提取溶剂,流速5ml/min,提取时间27秒。
本发明还有一个目的在于提供一种红花的加压微提取和离线检测羟基红花黄色素a的方法,具体操作为:
i.填装提取柱:红花药材粉碎,过60~80目筛,取1.0mg装于去除了填料的预柱芯中并用惰性材料硅藻土4.0~6.0mg进行填充,然后装入预柱套中置于液相色谱仪柱温箱中;
ii.加压微提取:柱温箱温度为55~65℃,以体积百分浓度25%的甲醇水溶液为提取溶剂,开启液相高压泵,流速3~5ml/min,提取时间20~120秒;收集流出液,混合均匀,即得到红花提取液;
iii.取步骤ii得到的红花提取液5μl,注入高效液相色谱仪中检测羟基红花黄色素a的含量。
优选的,上述步骤iii中的色谱条件为:
色谱柱:uplchsst3,100mm×2.1mm,1.8μm;
柱温:35℃;
流动相:0.01%甲酸(体积百分比浓度)水为a相,0.01%甲酸(体积百分比浓度)乙腈为b相,按照如下程序进行梯度洗脱:
0-5min,0%→23%b(体积百分比),余量为a;
5-8min,23%→100%b(体积百分比),余量为a;
流速:0.4ml/min;
检测器:光电二极管阵列检测器,检测波长为403nm。
还优选的,上述步骤iii还包括将对照品溶液5μl注入高效液相色谱仪中,其中所述对照品溶液通过如下方法制备:
称取羟基红花黄色素a标准品,加25%甲醇溶解配制成浓度为6.925mg/ml的储备溶液;取所述储备液用25%甲醇溶液稀释至羟基红花黄色素 a浓度为35μg/ml的标准液,即得。
此外,本发明还有一个目的在于提供一种加压微提取和在线检测的方法,包括将上述加压微提取设备和检测系统连接,使提取溶剂经所述加压设备进入所述提取柱中,流出液直接或经过处理后进入所述检测系统。
优选的,所述检测系统包括液相色谱分析柱和检测器。
优选的,所述加压微提取设备通过多通路阀门与所述液相色谱分析柱连接;提取时,使所述提取柱与所述液相色谱分析柱串联,开启所述加压装置后,提取溶剂流入所述提取柱,流出液中的化学成分在所述液相色谱分析柱前端富集;提取结束后,所述加压装置与所述液相色谱柱直接连通,使流动相直接进入所述液相色谱柱,对富集的化学成分进行洗脱和分离,洗脱液用检测器进行检测。
还优选的,所述加压微提取设备和所述检测系统之间还包括固相萃取小柱,加压提取液经过所述固相萃取小柱纯化后再进入所述检测系统。
作为一个优选的实施方式,本发明提供一种红花的加压微提取和在线检测的方法,具体的操作为:
i.填装提取柱:红花药材粉碎,过60~80目筛,取1.0mg装于去除了填料的预柱芯中并用惰性材料硅藻土4.0~6.0mg进行填充,然后装入预柱套中置于液相色谱仪柱温箱中;
ii.连接加压微提取设备和检测系统:所述提取柱、液相高压泵、分析色谱柱连接到六通阀上,所述分析色谱柱和检测器连接;
iii.加压微提取:切换六通阀,使所述液相高压泵、提取柱和分析色谱柱顺序串联;柱温箱温度为50~60℃,以体积百分浓度0.01%的甲酸水溶液为提取溶剂,开启液相高压泵,流速1ml/min,提取时间3分钟;
iv.在线检测:提取结束后,立即切换六通阀使所述液相高压泵和分析色谱柱直接连通,进行洗脱和检测。
优选的,所述步骤i中,硅藻土的填充量为6.0mg。
优选的,所述步骤ii中,柱温箱温度为55℃。
优选的,所述步骤iv中,色谱条件为:
色谱柱:waterscsh色谱柱,(150mm×4.6mm,3.5μm),
柱温:35℃,
流动相:以体积百分浓度0.01%的甲酸水溶液为a相,以体积百分浓度0.01%的甲酸乙腈溶液为b相;按照如下程序进行梯度洗脱:
0-3min,0%→12%b(体积百分比),余量为a,
3-20min,12%→30%b(体积百分比),余量为a,
23-26min,30%→100%b(体积百分比),余量为a;
流速:1ml/min。
还优选的,所述步骤iv中,所述检测器为质谱检测器,质谱检测条件为:
离子源:电雾化离子源,
检测模式:负离子模式检测,
离子喷射电压:-4500v,
温度:650℃,
源内气体1和气体2:都为氮气,压力皆为65psi,
气帘气体:氮气,压力为40.0psi,
扫描方式:多重反应检测。
作为另一个优选的实施方式,本发明提供一种肉苁蓉的加压微提取和在线检测的方法,具体的操作为:
i.填装提取柱:肉苁蓉药材粉碎,过60~80目筛,取1.0mg装于去除了填料的预柱芯中并用惰性材料正相硅胶4.0~6.0mg进行填充,然后装入预柱套中置于液相色谱仪柱温箱中;
ii.连接加压微提取设备和检测系统:所述提取柱、液相高压泵、c18色谱柱和固相萃取小柱连接到一个六通阀上,所述c18色谱柱、检测器和固相萃取小柱连接到另一个六通阀上;所述c18色谱柱和检测器连接;
iii.加压微提取:调节两个六通阀,使所述液相高压泵、提取柱固相萃取小柱顺序串联;柱温箱温度为55~65℃,以体积浓度0.1%的甲酸水溶液为提取溶剂,开启液相高压泵,流速2~4ml/min,提取时间60~180秒;
iv.在线检测:提取结束后,调节两个六通阀使所述液相高压泵、所述固相萃取小柱和分析色谱柱顺序串联,进行洗脱和检测。
优选的,所述步骤i中,正相硅胶的填充量为6.0mg。
优选的,所述步骤iii中,柱温箱温度为60℃。
优选的,所述步骤iii中,体积浓度0.1%的甲酸水溶液的流速为2ml/min,提取时间为180秒。
优选的,所述步骤iv中,色谱条件为:
柱温:室温,
流动相:以体积百分浓度0.1%的甲酸水溶液为a相,以乙腈溶液为b相;按照下述程序梯度洗脱:
0-32min,10%→30%b,余量为a;
流速:0.25ml/min,
检测器:光电二极管阵列检测器,检测波长278nm。
优选的,所述步骤iv中,还包括将对照品溶液10μl注射入所述高效液相色谱仪;所述对照品溶液通过如下方法制备:
分别称取肉苁蓉苷e、松果菊苷、毛蕊花糖苷、异毛蕊花糖苷、肉苁蓉苷c、2′-乙酰基金石蚕苷、异肉苁蓉苷c、管花苷b标准品,加dmso溶解配制成浓度均为4mg/ml的储备溶液;取所述各标准品储备液混合,用50%甲醇溶解稀释成各标准品浓度为400μg/ml的混合标准储备液,即得。
利用发明所述的加压微提取设备以及加压微提取和在线检测方法,可以广泛地应用于各种药用植物(包括中药材),进行微提取和成分检测,并不限于上述的红花和肉苁蓉。而且,根据药用植物所含成分的理化性质及提取、分析目的,可以灵活地采用合适的提取溶剂、洗脱溶剂、分析色谱柱以及检测器。
现有技术中,利用高效液相色谱法对药用植物(包括中药材)进行成分检测和分析时,供试品溶液通常采用加热回流提取、超声振荡提取或冷浸法,无论哪种方法,都需要消耗数克待测样品和数十至上百毫升的提取溶剂,耗费几十分钟到数小时,而实际分析仅需几十微升,剩余的供试品溶液都只能废弃。这样不仅造成待测样品和有机溶剂的浪费,而且容易污染环境。另外对于贵重中药材,比如虫草、红花等,由于分析一个样品需要消耗的药材量相对较大,使测试成本居高不下。
本发明提供的加压微提取设备以及基于其的加压微提取和在线检测方法,以已经得到广泛应用的高效液相色谱仪为基础,在高压和较高温度条件下,每次提取仅需要毫克级药材,几毫升提取溶剂,提取时间短则几十秒,长不过几分钟。而且以红花和肉苁蓉的提取实例看,提取效率高于现有技术的超声提取——提取的化学成分数量更多,各成分的含量更高。因此,本发明的方法实现了对包括中药材的药用植物快速、微量检测。相较现有技术,耗费的有机溶剂和样品量大幅减少,检测成本也相应降低。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
图1示出的是实施例3的红花提取液的液相色谱图,其中1号色谱峰为羟基红花黄色素a的吸收峰。
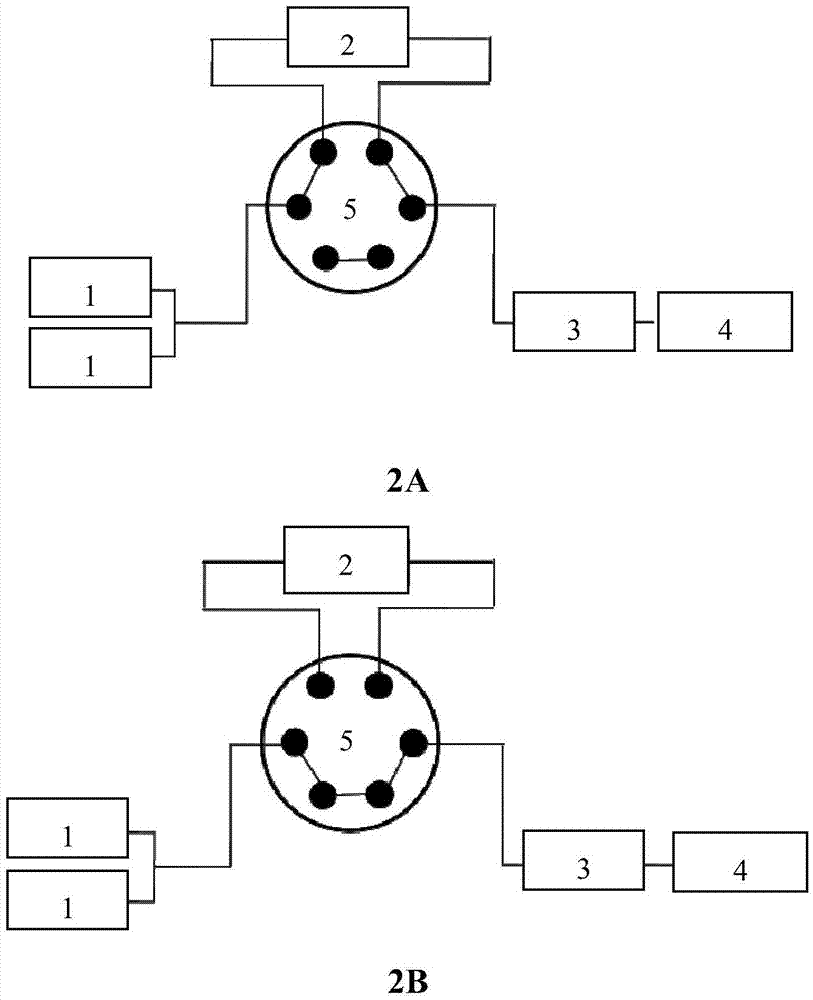
图2示出的是实施例5的加压微提取和在线检测设备结构示意图,其中图2a示出的是提取和富集时的装置连接情况,图2b示出的是分析是的装置连接情况。图中,1是高压输液泵,2是提取柱,3是分析柱,4是检测器,5是六通阀。
图3示出的是实施例5的红花不同提取方法得到的提取液的色谱图;其中a是红花经加压微提取得到的提取液在线检测得到的色谱图,b是红花经温浸提取得到的提取液的色谱图。
图4示出的是实施例5的红花经加压微提取得到的提取液和经温浸提取得到的提取液的色谱峰峰面积比较图,其中图4a示出c1-c21化合物的色谱峰面积比较结果,图4b示出c22-c43化合物的色谱峰面积比较结果。图中■代表加压微提取,□代表温浸提取。
图5示出的是实施例6的加压微提取和在线检测设备结构示意图,其中图5a示出的是提取和富集时的装置连接情况,图5b示出的是分析时的装置连接情况。图中,1是高压输液泵,2是提取柱,3是固相萃取小柱(spe柱),4是分析柱,5是检测器,6是六通阀。
图6示出的是实施例6利用加压微提取和在线检测方法得到的对照品溶液、荒漠肉苁蓉和管花肉苁蓉的色谱图,其中a是对照品溶液的色谱图,b是荒漠肉苁蓉的色谱图,c是管花肉苁蓉的色谱图。图中1号峰是肉苁蓉苷e的吸收峰,2号峰是松果菊苷的吸收峰,3号峰是毛蕊花糖苷的吸收峰,4号峰是异毛蕊花糖苷的吸收峰,5号峰是肉苁蓉苷c的吸收峰,6号峰是异肉苁蓉苷c的吸收峰,7号峰是2′-乙酰基金石蚕苷的吸收峰,8号峰是管花苷b的吸收峰。
具体实施方式
以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施 例中所用的药材原料、试剂材料等,如无特殊说明,均为市售购买产品。
其中,实验仪器:
色谱仪:1)岛津高效液相色谱仪(泵:lc-20adx2,自动进样器sil-20a,柱温箱cto-20a),超纯水净化系统,millipore公司;
2)watersacquityuplch-class系统,配有四元溶剂管理器(qsm)、样品管理器(sm-ftn)和光电二极管阵列检测器(pda),美国沃特世科技有限公司。
质谱仪:absciexqtrap4500型三重四级杆线性离子阱复合型串联质谱仪(配有离子喷雾离子源,analysis1.6.2数据处理系统,美国absciex公司)。
色谱柱:1)uplchsst3色谱柱(100mm×2.1mm,1.8μm),2)csh色谱柱(150mm×4.6mm,3.5μm),3)aceultracoresuperc18色谱柱(150mm×3.0mm,2.5μm)。
预柱套phenomenexsecurityguardtm,柱芯phenomenexcartridge(3.0mm×4mmi.d.)且去除rp-c18填料。
实验药材:
红花药材为菊科植物红花carthamustinctoriusl.的干燥花。肉苁蓉药材均为列当科植物荒漠肉苁蓉cistanchedeserticolay.c.ma或管花肉苁蓉cistanchetubulosa(schrenk)wight的干燥带鳞叶的肉质茎。
实验试剂与药品:
羟基红花黄色素a(hsya,批号:must-14101810,纯度hplc检测,>98%),购于成都曼斯特生物科技有限公司;
肉苁蓉苷e(cistanosidee)、松果菊苷(echinacoside)、毛蕊花糖苷(acteoside)、异毛蕊花糖苷(isoacteoside)、肉苁蓉苷c(cistanosidec)、2′-乙酰基金石蚕苷(2'-aceylpoliumoside)、异肉苁蓉苷c(isocistanosidec)、管花苷b(tubulosideb)由北京大学中药现代研究中心从荒漠肉苁蓉中提取分离,纯度大于95%。
液质纯乙腈、甲醇、甲酸,购于赛默飞世尔(中国)科技有限公司;硅藻土,化学纯,购于西陇化工股份有限公司;正相硅胶,100-200目,购于青岛海洋化工有限公司。
实施例1红花加压微提取研究和条件优化
1.基本条件
指标成分:羟基红花黄素a(hsya)
色谱仪:watersacquityuplch-class系统
色谱柱:uplchsst3色谱柱(100mm×2.1mm,1.8μm)
提取柱:将红花药材粉末(过60目筛)1.0mg装于预柱芯中并用惰性材料硅藻土进行填充,得到提取柱,后装入预柱套中置于柱温箱中。
对照品溶液制备:称取适量hsya标准品,加体积百分浓度25%的甲醇水溶液(以下简称“25%甲醇”)溶解配制成浓度为6.925mg/ml的储备溶液。取上述标准品储备液适量混合,用25%甲醇溶液稀释至化合物浓度为35μg/ml的标准液。
检测方式:采用uplc-pda系统进行hsya的含量测定
色谱条件:uplchsst3色谱柱(100mm×2.1mm,1.8μm);柱温:35℃;流动相:0.01%甲酸水(a)-0.01%甲酸乙腈(b),流速:0.4ml/min,梯度洗脱:0-5min,0%-23%b,5-8min,23%-100%b;检测波长403nm。
2.红花加压微提取及提取条件优化
将提取柱与色谱仪的高压泵连接,提取溶剂经高压泵输送至提取柱,收集流出液,取适量(5μl)注入uplc-pda系统进行分离和检测,记录hsya的色谱峰面积,并进行含量计算。
2.1提取溶剂
为了与超声提取具有可比性,加压微提取也采用体积百分比浓度25%的甲醇水溶液为提取溶剂。
2.2提取流速的考察
提取流速影响着提取的速度以及提取过程中的压力,是影响提取效率的重要因素。因此对不同的提取流速进行考察。以25%甲醇,60℃(柱温箱温度)为基本条件,分别考察了3ml/min(压力:2400psi)提取3min和5ml/min(压力:3600psi)提取1.8min的提取效率,结果见表1。
表1不同提取流速考察结果(n=3)
流速5ml/min,提取液中hsya的含量高,说明该流速下对hsya的提取效率更好,因此,优选提取流速为5ml/min。
2.3提取时间的考察
提取时间过短,样品不能完全提取,导致提取效率低;提取时间过长,溶剂消耗大,样品在高温状态下容易降解,同样导致提取效率低。在流速5ml/min,温度60℃条件下,考察了不同的提取时间:27秒(s)、54秒(s)和108秒(s)下的提取效率,结果见表2。
表2提取时间考察结果(n=3)
a:提取效率通过如下公式计算
提取效率=测得的hsya量/称取的药材量×100%(w/w%)
结果显示,提取时间27~54秒,提取效率基本不变,但是随着时间进一步延长,提取效率反而降低。因此,加压微提取只需提取27~54秒就可以达到完全提取的目的;且提取时间越短,所需试剂越少;故优选27秒。
2.4提取温度的考察
温度过低,化学成分溶出效率低;温度过高,hsya容易降解,也导致提取效率低。在提取流速为5ml/min,提取时间为27s的基本条件下,对50℃、55℃、60℃、65℃和70℃五个不同的提取温度进行考察,结果见表3。
表3提取温度考察结果(n=3)
表3的数据显示,红花在55~65℃进行提取,提取液中hsya的浓度都较高,其中55℃时最高。因此,提取温度优选为55~65℃,最优选为55℃。
2.5硅藻土装填量的考察
惰性材料的作用主要是分散样品微粒以及防止过细的样品微粒堵塞提取管的出口。因此考察了装填不同量的硅藻土的提取结果。结果见表4。
表4硅藻土装填量考察结果(n=3)
表4结果显示,显示硅藻土4mg或6mg对提取效率影响不大。但装填量越多,越紧实,样品分散程度越大,批间稳定性越好,故优选硅藻土的装填量为6.0mg。
2.6提取基质对提取效率的影响考察
制备空白提取柱:预柱芯不装填红花,仅装填6.0mg硅藻土。
将浓度为6.925mg/ml的对照品储备液2μl(相当于13.85μghsya)进样通过所述空白提取柱,检测hsya峰面积,记为“样品峰面积”。同时将浓度为6.925mg/ml的对照品储备液2μl直接进样至uplchsst3色谱柱,检测hsya的峰面积,记为“对照品峰面积”。结果见表5。
表5提取基质对提取效率的影响考察结果(n=6)
a:基质效应通过如下公式计算:
基质效应=样品峰面积/对照品峰面积×100%
表5结果显示,平均基质效应为96.50%,rsd值为4.50%;表明硅藻土不会影响在线加压溶剂微提取的效率。
2.7红花加压微提取优选条件
经过上述研究,建立了红花加压微提取的优选的工艺条件:
红花药材粉碎,过60~80目筛,取1.0mg装于去除了填料的预柱芯中并用惰性材料硅藻土4.0~6.0mg进行填充,然后装入预柱套中置于液相色谱仪柱温箱中;柱温箱温度为55~65℃,以体积浓度25%的甲醇水溶液为提取溶剂,流速3~5ml/min,提取时间20~60秒;收集流出液。
红花加压微提取最优选的工艺条件为:
红花药材粉碎,过60目筛,取1.0mg装于去除了填料的预柱芯中并用惰性材料硅藻土6.0mg进行填充,然后装入预柱套中置于液相色谱仪柱温箱中;柱温箱温度为55℃,以体积浓度25%的甲醇水溶液为提取溶剂,流速5ml/min,提取时间27秒;收集流出液。
实施例2红花加压微提取方法学考察
以实施例1建立的最优选的红花加压微提取条件,以及实施例1所述的色谱条件下,进行方法学考察。
1.线性范围考察
以实施例1配备的对照品标准液分别进样0.1、0.2、0.5、1、2、5μl, 记录相应的色谱峰面积。以对照品峰面积y对进样量x(μg)进行线性回归。回归方程为:y=5196429.6x-3599.9(r2=1),结果表明进样量在3.5-175ng线性关系良好。
2.精密度考察
取红花药材粉末约1.0mg,精密称定,装于预柱芯中并用硅藻土6.0mg进行填充后,装入预柱套中,得到提取柱。将提取柱置于柱温箱中,按实施例1建立的最优选的红花药材加压微提取的条件进行提取后,提取液进样5μl,连续进样6次,记录色谱峰面积。结果见表6。
表6精密度考察结果(n=6)
表6的数据表明仪器精密度良好,rsd值为0.2%,符合分析方法的要求。
3.重复性考察
取红花药材粉末约1.0mg,精密称定,装于预柱芯中并用硅藻土6.0mg进行填充后,装入预柱套中,得到提取柱。平行制备6个样品。将提取柱置于柱温箱中,按实施例1建立的最优选的红花药材加压微提取的条件进行提取后,进样5μl,计算hsya的含量。结果见表7。
表7重复性考察结果(n=6)
表7结果显示rsd<5%,表明重复性良好,符合分析方法的要求。
4.加样回收率考察
取红花药材粉末约1.0mg,精密称定,装于预柱芯中并用硅藻土6.0mg进行填充,装入预柱套中,平行6个样品。将提取柱置于柱温箱中,将浓度为6.925mg/ml的对照品储备液2μl(相当于13.85μghsya)注射进样于提取柱内,然后按实施例1建立的最优选的红花药材加压微提取的条件进行提取后,收集提取液,进样5μl,测定并记录峰面积,计算hsya的加样回收率,结果见表8。
表8加样回收率考察结果(n=6)
表8数据显示hsya的加样回收率为95.71%,rsd值为3.64%,符合分析方法的要求。
本实施例的结果显示,本发明建立的红花加压微提取方法稳定性、重现性好。
实施例3一种红花加压微提取方法
红花药材粉碎,过60目筛,取1.0mg装于去除了填料的预柱芯中并用惰性材料硅藻土6.0mg进行填充,然后装入预柱套中置于液相色谱仪柱温箱中;柱温箱温度为55℃,以体积浓度25%的甲醇水溶液为提取溶剂,流速5ml/min,提取时间27秒;收集流出液,即为红花提取液。
采用实施例1的检测方式和色谱条件,得到所述提取液的液相色谱图,见图1。
实施例4本发明的加压微提取方法和传统提取方法的比较
1.供试品溶液的制备
a.红花药材在线加压溶剂微提取:将红花药材粉末1.0mg装于预柱芯中并用惰性材料硅藻土6.0mg进行填充,后装入预柱套中,将提取柱置于柱温箱中,以25%甲醇为提取溶剂,提取温度55℃,流速5ml/min(压力:3600psi),加压提取27秒。收集提取液,取5μl注入uplc中分析,记录相应的色谱峰面积并计算提取效率。
b.红花药材超声提取:根据《中国药典》(2010版)中红花药材中hsya含量测定的方法进行提取,即取红花药材粉末(过60目筛)约0.1g,精密称定后置具塞锥形瓶中,精密加入25%甲醇12.5ml,称定重量,超声处理(功率300w,频率5khz)40分钟,放冷,再称定重量,用25%甲醇补足减失的重量,摇匀,滤过,取续滤液0.5μl注入uplc中分析,记录相应的色谱 峰面积并计算提取效率。
2.色谱分析条件:
同实施例1的色谱条件。
3.测定结果:见表9,同时将两种提取方法的样品和溶剂用量、提取时间等也列于表9,以便比较。
表9在线加压溶剂微提取与超声提取的比较(n=3)
表9的数据显示,本发明的加压微提取方法与超声提取法相比,提取率提高,并且能明显的缩短提取时间、减少样品和溶剂的用量,具有显著的优势。
实施例5一种红花加压微提取和在线检测的方法
1.加压微提取和在线检测设备
按照图2所示建立加压微提取和在线检测设备,其中1是高压输液泵,2是提取柱,3是分析柱,4是检测器,5是六通阀。
2.检测条件:
2.1色谱条件
分析柱:waterscsh色谱柱(150mm×4.6mm,3.5μm,货号:186005270);
柱温:室温;
流动相:0.01%甲酸水(a)-0.01%甲酸乙腈(b),室温,流速:1ml/min;梯度洗脱程序见表10。
表10在线加压溶剂微萃取对红花中微量成分提取并洗脱流动相程序
2.2检测器:
absciexqtrap4500型三重四级杆线性离子阱复合型串联质谱仪,采用qtrap4500的多反应检测模式(mrm)对红花中的微量成分进行检测,相应的质谱条件如下:离子源为电雾化离子源;负离子模式检测,离子喷射电压为-4500v;温度为650℃;源内气体1和气体2:氮气压力为皆为65psi;气帘气体:氮气压力为40.0psi;扫描方式为多重反应检测。
3.提取和检测
将红花药材粉末(过60目筛)1.0mg装于预柱芯中并用惰性材料硅藻土6.0mg进行填充,将填充好的预柱芯装入保护柱套中置于柱温箱中,将六通阀调节至图2a所示的位置,使提取装置与分析柱串联。提取溶剂为0.01%甲酸水,流速为1ml/min,柱温箱提取时的温度为55℃。样品经过高温、加压提取,并在分析柱上富集。提取完成后,将六通阀调节至图2b所示的位置,使流动相直接经过分析柱,对保留在分析柱前端的成分进行洗脱分离,并注入质谱中进行分析。色谱图见图3,记录相应峰面积,结果见图4a和图4b。
4.红花药材温浸提取
取红花药材粉末(过60目筛)约0.5g,精密称定后置具塞锥形瓶中,精密加入0.01%甲酸水溶液5.0ml,称定重量,于55℃水浴温度下提取60分钟,放冷,再称定重量,用0.01%甲酸水溶液补足减失的重量,摇匀,滤过,取续滤液10μl(折合至上样量为1.0mg,与加压微提取的上样量相同)注入色谱仪,洗脱及检测方式同本实施例的“加压微提取和在线检测”。色谱图见图3,记录峰面积,结果见图4a和图4b。
5.结论:
通过比较本发明的加压微提取和在线检测法和温浸提取法得到的提取液的相应吸收峰的峰面积,可以看出:在同样的提取溶剂(0.01%甲酸水溶液)、提取温度(55℃)以及检测条件下,本发明的加压微提取方法得到的提取液中各成分的吸收峰面积均大于温浸法得到的提取液的相应成分。表明本发明的加压微提取方法不仅提高了提取效率,而且节约了样品及溶剂,更适合于红花中微量成分的提取和检测。
实施例6一种肉苁蓉加压微提取和在线检测方法
1.加压微提取和在线检测设备
按照图5所示建立加压微提取和在线检测设备,其中1是高压输液泵,2是提取柱,3是固相萃取小柱(spe柱),4是分析柱,5是检测器,6是六通阀。
加压微提取和在线检测的工作过程是:提取时,两个六通阀6位置如图5a所示,提取柱2(此时提取柱置于柱温箱中)与spe柱3串联,样品经过高温加压提取,并在spe柱3上富集。提取结束后,两个六通阀位置切换至如图5b所示,spe柱3与分析柱4串联,保留在spe柱3上的成分被反冲到分析柱4进行分离,并注入检测器5中进行检测,记录色谱图并进行相应的计算;此时可以对提取柱2进行样品更换。
2.测试样品制备
2.1提取柱
称取肉苁蓉药材(过四号筛)1mg装入预柱芯中,并用正相硅胶6mg填充,将预柱芯装入预柱套中,待用。提取时将提取柱置于柱温箱中。
2.2对照品溶液制备
称取适量肉苁蓉苷e、松果菊苷、毛蕊花糖苷、异毛蕊花糖苷、肉苁蓉苷c、2′-乙酰基金石蚕苷、异肉苁蓉苷c、管花苷b,加dmso溶解配制成各标准品浓度为4mg/ml的储备溶液。取上述标准品储备液适量混合,用50%甲醇溶解稀释成各标准品浓度为400μg/ml的混合标准储备液,备用。
3.提取和检测条件:
3.1提取条件
提取溶剂:0.1%甲酸水溶液
提取温度:60℃
流速:2ml/min
3.2检测条件
分析柱:aceultracoresuperc18色谱柱(规格:150mm×3.0mm,2.5μm)(货号:core-25a-1002u);
检测波长:278nm;
柱温:室温;
流动相:0.1%甲酸水(a)-乙腈(b),室温;梯度洗脱程序见表11。
表11肉苁蓉加压微提取-固相萃取-在线检测流动相程序
4.方法学考察
4.1检测限和定量限
仅用洗净的正相硅胶填充预柱芯,置于预柱套内,得到空白提取柱。将混合标准储备溶液用50%甲醇不断稀释并上样于空白提取柱,按照加压微提取和在线检测的工作过程进行测定,分别以信噪比3和10为标准确定各化合物的检测线和定量限。
结果见表12。
4.2标准曲线
仅用洗净的正相硅胶填充预柱芯,置于预柱套内,得到空白提取柱。取混合标准储备液适量,用50%甲醇溶解稀释,制成各标准品浓度分别为200μg/ml、100μg/ml、80μg/ml、50μg/ml、25μg/ml、10μg/ml、5μg/ml、2.5μg/ml、1μg/ml。按照加压微提取和在线检测的工作过程进行测定,以目标化合物峰面积为纵坐标,浓度为横坐标,绘制标准曲线,得线性回归方程。
结果:见表12。
表12标准品的线性方程、检测限和定量限测定结果
表12的数据显示,各标准品化合物在检测限和定量限浓度范围内线性关系良好,本实施例的方法灵敏度高。
4.3精密度
a、日内精密度:仅用正相硅胶填充预柱芯,置于预柱套内,得到空白提取柱。精密吸取高(80μg/ml)、中(25μg/ml)、低(5μg/ml)三个浓度的混合标准溶液,分别连续进样六次。按照加压微提取和在线检测的工作过程进行测定,记录各化合物的峰面积,并计算相应的rsd值。
b、日间精密度:用正相硅胶填充柱芯,置于预柱套内。精密吸取高(80μg/ml)、中(25μg/ml)、低(5μg/ml)三个浓度的混合标准溶液,分别连续三天进样,每天进样三次。按照加压微提取和在线检测的工作过程进行测定,记录各化合物的峰面积,并计算相应的rsd值。
结果:见表13。
表13日内精密度(n=6)和日间精密度(n=9)测定结果
表13的数据表明,本方法精密度良好。
4.4重复性
称取约1mg荒漠肉苁蓉样品(过四号筛)装于预柱芯中,按照加压微提取和在线检测的工作过程进行测定,进样并记录保留时间和峰面积,并计算保留时间和各化合物在样品中含量的rsd值。
结果:见表14。
4.5加样回收率试验
称取约0.5mg荒漠肉苁蓉样品(过四号筛)装于预柱芯中,取高(80μg/ml)、中(25μg/ml)、低(5μg/ml)三个浓度的混合标准品溶液,根据测得量和加入量计算各化合物的回收率。
结果:见表14。
表148个化合物的回收率(n=3)和重复性(n=5)
表14的数据显示,本方法重复性良好,加样回收率结果良好。说明本方法准确度良好。
5.含量测定
分别取不同产地和批次的荒漠肉苁蓉和管花肉苁蓉1mg,分别装入预柱芯内,填充正相硅胶6mg后装入预柱套内,得到提取柱,置于柱温箱中,按照图6所示建立加压微提取和在线检测装置。提取溶剂为0.1%甲酸水溶液,提取温度60℃,提取流速2ml/min。检测条件如3.2项下所述。
提取时,两个六通阀6位置如图5a所示,提取柱2与spe柱3串联,样品经过高温加压提取,并在spe柱3上富集。提取结束后,两个六通阀位置切换至如图5b所示,spe柱3与分析柱4串联,保留在spe柱3上的成分被反冲到分析柱4进行分离,并注入检测器5中进行检测,记录色谱图并进行相应的计算。
对照品溶液、荒漠肉苁蓉和管花肉苁蓉的色谱图见图6(a-c)。含量测定结果见表15。
表15肉苁蓉含量测定结果(μg/mg,n=1)
表中1-8号化合物分别是肉苁蓉苷e(1),松果菊苷(2),毛蕊花糖苷(3),异毛蕊花糖苷(4),肉苁蓉苷c(5),异肉苁蓉苷c(6),2’-乙酰基金石蚕苷(7)和管花苷b(8)。
6.本实施例方法与超声提取法比较
6.1肉苁蓉超声提取
即取荒漠肉苁蓉(过四号筛)约0.5g,精密称定后置具塞锥形瓶中,精密加入50%甲醇50ml,称定重量,超声处理(功率300w,频率5khz)40分钟,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,取续滤液10μl注入uplc中分析,记录相应的色谱峰面积并计算提取效率。结果见表16。
表16在线加压溶剂微提取与超声提取的比较(n=5)
从提取溶剂、溶剂用量、提取时间、药材量以及提取效率等方面进行比较的结果显示,本发明的肉苁蓉加压微提取方法能够实现更高的提取率,其在使用水溶液的条件下能够达到有机溶剂(50%甲醇)对各化合物的提取效果,并且能明显地缩短提取时间、减少样品和溶剂的用量,无论在经济效益和环保方面都具有显著的优势。
本实施例提供的加压微提取和在线检测方法,能够对肉苁蓉进行快速、微量、准确的定性和定量检测。此外利用在线spe小柱对样品进行富集,在不需要使用有机溶剂进行提取的情况下,对极性较低的化合物仍达到了更高的提取效率,进一步说明该方法比传统的提取方式更为环保。
以上对本发明具体实施方式的描述并不限制本发明,本领域技术人员可以根据本发明做出各种改变或变形,只要不脱离本发明的精神,均应属于本发明所附权利要求的范围。
- 还没有人留言评论。精彩留言会获得点赞!