用于检测抗药物抗体的存在或抗药物抗体的量的测定的制作方法
本申请要求2014年2月11日提交的美国临时专利申请号61/938,556的权益,其整体内容在此处通过提述并入。
技术领域
本发明涉及用于检测样品中抗药物抗体的存在的方法和试剂盒,且更具体地涉及样品中药物存在下的用于检测抗药物抗体的方法和试剂盒。
背景
生物治疗剂(例如生物剂如蛋白、肽、核苷酸等)的引入已为疾病如炎性肠疾病、强直性脊柱炎、多发性硬化和类风湿性关节炎的治疗给出了主要推动。在很多情况中,已证明这些生物剂在临床实践中十分成功。已知生物剂包括治疗性抗体具有免疫原性的潜力,而对患者施用治疗性蛋白可诱导免疫响应,导致形成抗药物抗体(“ADA”)。这种ADA可减少治疗性蛋白的有效性。例如,其可结合或/和中和治疗性蛋白,导致药物药代动力学或药效动力学的改变,其改变药物效力。ADA可引起严重的副作用,包括过敏反应、通过中和抗体(NAb)的针对内源性蛋白的交叉反应性和补体激活。对于数种可用于治疗类风湿性关节炎(阿达木单抗和英利昔单抗)、克罗恩病(英利昔单抗)、多发性硬化(那他珠单抗和阿仑单抗)和斑块状银屑病(阿达木单抗)的单克隆抗体,已描述了ADA的产生。在一些患者中,由这些治疗性蛋白提供的临床效益因为ADA的形成而随时间减少。免疫原性风险评估对于理解药物诱导的ADA的频率和严重性至关重要。已报导Nab与引起耗竭综合征的内源性蛋白具有交叉反应(促红细胞生成素)。
随着批准用于临床使用的治疗性蛋白数量的增加,这些产品的免疫原性对于临床医生、制造商和监管机构已变得翔实。已明确了解一些底物会影响免疫测定(或配体结合测定)中分析物的检测或量化。这些干扰因素包括但不限于消极影响测定的特异性、精确性和敏感性的循环的药物。将减少ADA测定“药物耐性”的“药物干扰”认作是对于免疫原性评估(以监测ADA作为患者药物临床安全性和效力的监测的一部分)的主要技术挑战。
尽管上述方法证明了药物耐性的一定改善,但相比无药物的ADA检测,其不能维持敏感性和相对的精确性,因此具有假阴性的风险且低报在治疗的患者中ADA的发生率和效价。尽管工业监管指导文件和白皮书推荐的敏感性为250-500ng/mL[Shankar G,Devanarayan V,Amaravadi L等:Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products.Journal of Pharmaceutical and Biomedical Analysis 48(5),1267-1281(2008);Mire-Sluis AR,Barrett YC,Devanarayan V等:Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products.Journal of Immunological Methods 289(1-2),1-16(2004)],药物耐性有时在无任何接受标准下进行评估,然后写出临床规程来指示抗体测量前的长洗出期以允许药物清除并避免由于药物干扰导致的假阴性结果。该方法并不合意,这是由于在早期的时间点尤其是在具有长半衰期药物和/或多次给药方案的情况中,将存在错过ADA评估的风险且洗出期方案并不可行。一些基于非配体结合的方法如质谱已在ADA干扰存在下用于PK评估,预期的测定敏感性是不能接受的且需要基于配体的结合来富集分析物,这造成相似的挑战。
多种测定形式已成功用于检测ADA,包括ELISA(直接的、间接的和桥接的)、放射免疫测定,电化学发光和表面等离子体共振。然而这些测定的开发经常由于药物存在引起的干扰而复杂化。长久以来已认识到在配体结合测定中分析干扰的挑战。随着长效单克隆抗体疗法的到来,对于在药物存在下检测ADA的特定技术的需求引起了特别关注。目前使用的最广泛采用的方式仍具有时间、敏感性和精确性的限制。因此,本领域对于更精确和可重复地检测样品如生物样品中ADA存在的方法和试剂盒存在需求。
概述
本发明至少部分基于新的测定方法的发现,所述方法可有效用于减少或消除由ADA检测中的药物或靶的干扰所引起的问题。具体地,本发明是基于新的ADA测定的建立,所述测定包括下列示例性的步骤。首先,将过量的药物材料添加至包含潜在ADA(游离的ADA和ADA/药物复合物)的样品以结合全部残留的游离ADA,形成药物/ADA复合物。第二,使用聚乙二醇沉淀这些复合物。第三,系列洗涤以去除血清蛋白和免疫球蛋白后,用溶液重构最终沉淀(药物/ADA复合物)以解离所述复合物然后以足够允许涂覆全部解离的游离药物和游离ADA的时间涂覆在大的表面(在保持药物和ADA分离的条件下)或基质(例如具有高涂覆容量的高结合碳板)上。第四,随后使用标记的药物实施总ADA水平的特定检测。因此,本发明涉及用于确定样品(例如生物样品)中ADA存在或ADA的量的方法、组合物和试剂盒。
在一个实施方案中,用酸溶液重构最终沉淀(药物/ADA复合物)以解离所述复合物然后以足够允许涂覆全部解离的游离药物和游离ADA的时间涂覆在大的表面(在保持药物和ADA分离的酸性条件下)或基质(例如具有高涂覆容量的高结合碳板)上。所述酸性环境防止所述复合物在固定至基质表面上时再次形成。当使用酸性溶液解离复合物时,可将测定称为PandA(PEG和Acid(酸))测定。
在另一实施方案中,用碱溶液重构最终沉淀(药物/ADA复合物)以解离所述复合物然后以足够允许涂覆全部解离的游离药物和游离ADA的时间涂覆在大的表面(在保持药物和ADA分离的碱性条件下)或基质(例如具有高涂覆容量的高结合碳板)上。所述碱性环境防止所述复合物在固定至基质表面上时再次形成。
酸性或碱性溶液的选择将取决于药物(例如生物药物)的参数,如pI,或一些共轭键的存在,且所述选择对药物的完整性和结构会具有最小的影响。
在一些实施方案中,最初的酸或碱添加后的温育步骤可在22℃、23℃、25℃、27℃、30℃、32℃、35℃、37℃、39℃或更高实施。
在一些实施方案中,最终的沉淀步骤后,可将每个样品进一步稀释至例如1:20、1:25、1:30、1:40、1:50或1:60的最终样品稀释度。
一方面,本公开内容提供了用于确定样品中ADA存在或不存在的方法,所述方法包括使所述样品与结合ADA以形成药物/ADA复合物的过量的药物接触;使所述药物/ADA复合物与聚乙二醇(PEG)接触以形成包含药物/ADA复合物的沉淀;使所述沉淀与溶液接触以解离所述药物/ADA复合物;将所述解离的ADA固定于表面和/或基质上;和确定所述ADA的存在或量。另一方面,所述确定步骤包括:使所述固定的ADA与缀合有可检测的标记物的药物接触;和确定所述可检测的标记物的存在或量,以由此确定所述样品中ADA的存在或量(效价)。
在一些实施方案中,用于确定样品中ADA的存在或不存在的方法进一步包括在所述确定步骤的部分之前、之后确定样品中ADA的量。
在一些实施方案中,用于确定生物样品中ADA的存在或不存在的方法进一步包括在所述固定步骤后处理(例如洗涤)所述支持物以去除未结合的药物。
在其他实施方案中,用于确定生物样品中ADA存在或不存在的方法进一步包括在将样品与PEG接触后洗涤沉淀。
在其他实施方案中,所述方法进一步包括在将ADA固定在基质上的步骤之前、之后或在其间将所述药物固定在基质上。
另一方面,本文提供了用于减少药物测定(例如药物PK测定、药物定量测定或药物效力测定)中由于样品中ADA的存在导致的干扰,所述方法包括使样品与过量的ADA接触以饱和游离的药物并形成药物/ADA复合物,使所述药物/ADA复合物与聚乙二醇(PEG)接触,以由此形成包含药物/ADA复合物的沉淀,使所述沉淀与溶液接触以解离所述药物/ADA复合物,在酸性条件下将所述解离的药物固定在基质上,和使用用于药物的特定检测剂实施药物测定,以由此减少来自所述ADA的干扰。所述药物测定可以是例如药物量化测定、药物PK测定或药物效力测定。
在一些实施方案中,用于减少药物测定中由于ADA的存在所致的干扰的方法进一步包括使用以可检测的标记物标记的抗独特型抗体确定所述样品中所述药物的存在或不存在或量。
在一些实施方案中,本文公开的方法进一步包括在样品与过量的药物接触前将其稀释。例如,在样品与过量的药物接触前将样品稀释1:2、1:5、1:10、1:20倍。
在其他实施方案中,样品包括生物样品,其中所述生物样品包括选自下组的材料:体液、粘液分泌物、唾液、血液、全血、血浆或血清。在一些实施方案中,所述样品包含药物。
在其他实施方案中,药物包含抗体或其片段,双亲和性抗体、双抗体、多域生物剂(如抗体药物缀合物)、核酸(siRNA、反义寡核苷酸、基因疗法药物)、肽或多肽(天然的或修饰的)、肽模拟物、糖、脂质,或有机或无机小分子化合物,或其任意组合。在一些实施方案中,药物包含治疗性抗体、蛋白治疗剂、酶、工程化的结合蛋白、工程化的抗体样蛋白、融合蛋白、支架蛋白或其任意组合。当药物包含抗体或其片段时,抗体可以是鼠抗体、人抗体、人源化抗体或嵌合抗体。在一些实施方案中,药物是经修饰以与未经修饰形式的相同药物相比呈现较少免疫原性的药物(即药物已经修饰以具有较少免疫原性)。
在一些实施方案中,基质包含碳表面、玻璃表面、二氧化硅表面、金属表面、聚合材料、含有金属或化学涂层的表面、膜、珠(例如微珠)、多孔聚合物基质或包含纤维素纤维的基质,或其任意组合。所述基质可包含聚合材料,其中所述聚合材料选自下组:聚苯乙烯、聚氯乙烯、聚丙烯、聚乙烯、聚酰胺和聚碳酸酯。
在一些实施方案中,基质包含多孔碳表面。在一个实施方案中,基质是高结合碳板。
在一些实施方案中,基质包含具有高涂覆容量的大表面。包含具有高涂覆容量的大表面的基质包括例如高结合碳板(例如MSD(Meso ScaleRockville Maryland)。
在一些实施方案中,提供的方法包括使药物/ADA复合物与聚乙二醇(PEG)接触以形成包含药物/ADA复合物的沉淀。PEG包含至少一种具有1,000-40,000道尔顿的PEG化合物,包括例如至少一种选自下组的PEG化合物:PEG1000、PEG1450、PEG3000、PEG6000、PEG8000、PEG10000、PEG14000、PEG15000、PEG20000、PEG250000、PEG30000、PEG35000和PEG40000。
在一个或多个实施方案中,所述样品与下述浓度的PEG接触:约0.1%至约10.0%、约0.2%至约7.0%,约0.5%至约6.0%,约0.5%至约5.0%,约1.5%至约5.5%,约2.0%至约5.0%,约3.0%至约4.5%,约3.5%至约4.0%,约1.0%至约2.5%,约1.2%至约1.5%,或约0.1%、0.2%、0.5%、1.0%、1.2%、1.5%、2.0%、2.5%、3.0%、3.5%、4.0%、4.5%、5.0%、5.5%、6.0%、6.5%、7.0%、7.5%、8.0%、8.5%、9.0%、9.5%或约10.0%PEG。
在其他实施方案中,所述方法包括使包含ADA和/或ADA/药物复合物的沉淀与酸溶液接触。所述酸溶液可包含有机酸、无机酸或其混合物。一些方面,所述酸溶液包含选自下组的酸:柠檬酸、异柠檬酸、谷氨酸、乙酸、乳酸、甲酸、草酸、尿酸、三氟乙酸、苯磺酸、氨基甲磺酸、樟脑-10-磺酸、氯乙酸、溴乙酸、碘乙酸、丙酸、丁酸、甘油酸、琥珀酸、苹果酸、天冬氨酸、盐酸、硝酸、磷酸、硫酸、硼酸、氢氟酸、氢溴酸,及其任意组合。在示例性的实施方案中,所述酸溶液包含乙酸。对于包括使含有ADA和/或ADA/药物复合物的沉淀与酸溶液接触的方法,将所述沉淀与约0.1M至约5M的浓度的酸接触。
在其他实施方案中,所述方法包括使含有ADA和/或ADA/药物复合物的沉淀与碱溶液接触。所述碱溶液可包括有机碱、无机碱或其混合物。一些方面,所述碱溶液包含选自下组的碱:尿素、氢氧化钠、氢氧化铷、氢氧化铯、氢氧化钙、氢氧化锶、氢氧化钡、氢氧化锌、氢氧化锂、丙酮、甲胺和氨。对于包括使含有ADA和/或ADA/药物复合物的沉淀与碱溶液接触的方法,将所述沉淀与约0.1M至约1M的浓度的碱接触。
一些方面,本公开内容提供抗体、抗药物抗体和用可检测的标记物标记(即与其缀合)的药物。可检测的标记物包括选自下组的标记物:半抗原、放射性同位素、酶、荧光标记物、化学发光标记物和电化学发光标记物、结合对的第一成员,及用于酶检测反应的底物。在一个实施方案中,可检测的标记物包括含有标记的电化学发光标记物。
在一些实施方案中,可检测的标记物包含荧光团,其中所述荧光团选自下组:绿色荧光蛋白,蓝色荧光蛋白、红色荧光蛋白、荧光素、荧光素5-异硫氰酸酯(FITC)、花青染料(Cy3、Cy3.5、Cy5、Cy5.5、Cy7)、Bodipy染料(Invitrogen)和/或Alexa Fluor染料(Invitrogen)、丹酰、丹磺酰氯(DNS-C1)、5-(碘乙酰胺)荧光素(5-IAF、6-丙烯酰-2-二甲氨基萘(Acrylodan)、7-硝基苯并-2-氧杂-1,3,-二唑-4-基氯化物(NBD-Cl)、溴化乙锭、萤光黄、罗丹明染料(5-羧基罗丹明6G氢氯化物、丽丝胺罗丹明B磺酰氯、罗丹明-B-异硫氰酸酯(RITC(罗丹明-B-异硫氰酸酯)、罗丹明800);四甲基罗丹明5-(和6-)异硫氰酸酯(TRITC))、Texas RedTM、磺酰氯、萘磺酸包括但不限于1-苯胺萘-8-磺酸(ANS)和6-(对-甲苯胺基)萘-e-2-磺酸(TNS)、氨茴酰脂肪酸、DPH、十八碳四烯酸、TMA-DPH、芴基脂肪酸、荧光素-磷脂酰乙醇胺、Texas红-磷脂酰乙醇胺、芘基-磷脂酰胆碱、芴基-磷脂酰胆碱、部花青540、苯乙烯基萘、3,3'二丙基硫代二碳青色素(diS-C3-(5))、4-(对二戊基氨基苯乙烯基)-l-甲基吡啶盐(di-5-ASP)、Cy-3碘乙酰胺、Cy-5-N-羟基琥珀酰亚胺、Cy-7-异硫氰酸酯、IR-125、噻唑橙、天青B、尼罗蓝、铝酞菁、噁嗪1,4',6-二脒-2-苯基吲哚。(DAPI)、Hoechst 33342、TOTO、吖啶橙、乙锭同型二聚体、N(乙氧羰基甲基)-6-甲氧基喹啉(MQAE)、呋喃-2、钙绿、羧基SNARF-6、BAPTA、香豆素、Phytofiuors和Coronene(晕苯)。
在一些实施方案中,可检测的标记物包括催化颜色改变反应的酶,其包括选自下组的酶:碱性磷酸酶、β-半乳糖苷酶、辣根过氧化物酶、脲酶和β-内酰胺酶和葡萄糖氧化酶。
在一些实施方案中,可检测的标记物包括结合对的第一成员或结合对的第二成员,其中所述结合对选自下组:生物素/链霉亲和素、生物素/抗生物素蛋白、生物素/中性抗生物素蛋白、生物素/凯普亲和素、表位/抗体、蛋白A/免疫球蛋白、蛋白G/免疫球蛋白、蛋白L/免疫球蛋白、GST/谷胱甘肽、His-标签/镍、抗原/抗体、FLAG/M1抗体、麦芽糖结合蛋白/麦芽糖、钙调蛋白结合蛋白/钙调蛋白、酶-酶底物和受体-配体结合对。
在一些实施方案中,可检测的标记物包括结合对的第一成员;且结合对的第二成员与酶、抗体表位、抗原、荧光团、放射性同位素、纳米颗粒、第二结合对的成员和金属螯合物缀合。
在其他实施方案中,可检测的标记物包括结合对的第一成员,其中所述结合对的第一成员是生物素且所述结合对的第二成员选自下组:链霉亲和素、抗生物素蛋白、中性抗生物素蛋白和凯普亲和素,且结合对的第二成员与酶缀合。
取决于待检测的样品数目,本文提供的方法可在人工或自动化的仪器平台上实施。
“抗药物抗体”或“ADA”是特异性地与药物的任何区结合的抗体。例如,抗药物抗体可以是抗体或其片段,所述抗体或其片段可针对药物抗体的任何区,例如可变域、恒定域或抗体的糖结构。这种抗药物抗体可在药物疗法过程中由于患者的免疫原性反应发生。ADA可以是任何人免疫球蛋白同种型(例如IgM、IgE、IgA、IgG、IgD)或IgG亚类(IgG1、2、3和4)之一。ADA包括来自任何动物来源的ADA,包括例如人或非人动物(例如兽医)来源。
就本说明书而言,术语“NAb”或“中和抗体”指与内源性产生的分子结合的抗体,例如抗体、核酸、肽、多肽、肽模拟物、糖或脂质。例如Nab可以是内源性产生的蛋白,如例如促红细胞生成素或胰岛素。Nab可以或可以不减少(例如中和)内源性产生的分子的至少一种生物活性。
例如,在一些方面,本公开内容提供了用于确定样品中Nab存在或不存在的方法,所述方法包括使样品与结合NAb以形成抗原/NAb复合物的过量的抗原接触;使抗原/NAb复合物与聚乙二醇(PEG)接触以形成包含抗原/NAb复合物的沉淀,使所述沉淀与溶液接触以解离抗原/NAb复合物,将解离的Nab固定在表面和/或基质上,并确定所述Nab的存在或量。在另一方面,确定步骤包括使固定的Nab与结合Nab并缀合有可检测的标记物的抗原接触,并确定所述可检测的标记物的存在或量以由此确定样品中Nab的存在或量(效价)。
在本发明的上下文中,术语“患者”指任何具有疾病或疑似具有疾病的受试者,优选哺乳动物,且更优选人。如本文使用的术语“受试者”指任何动物(例如人或非人动物受试者)。在一些实例中,受试者是哺乳动物。在一些实例中,如本文使用的术语“受试者”指人(例如男性、女性或儿童)。在一些实例中,如本文使用的术语“受试者”指动物模型研究的实验室动物。
如本文使用的术语“生物样品”或“样品”指获得自或源自患者的样品,所述样品包含源自患者的免疫球蛋白且可因此可称作免疫球蛋白样品。举例来说,生物样品包含选自下组的材料:体液、血液、全血、血浆、血清、粘液分泌物、唾液、脑脊液(CSF)、支气管肺泡灌洗液(BALF)、眼部流体(例如玻璃体液、房水)、淋巴液、淋巴结组织、脾组织、骨髓和源自一种或多种这些组织的免疫球蛋白富集组分。在一些实施方案中,样品是或包含血清或为源自血清或血液的免疫球蛋白富含级分。样品是或可源自(获得自)体液或身体组织。在一些实施方案中,样品获得自已暴露至(如反复暴露至相同药物)药物的受试者。在其他实施方案中,所述样品获得自近期未暴露至药物的受试者,或获得自计划施用药物前的受试者。
如本文使用的术语“基质”指可结合任何ADA或药物材料(例如抗体、核酸、肽、多肽、肽模拟物、糖、脂质或有机或无机小分子化合物)的材料或大分子复合物。基质的组成或表面应允许ADA或药物材料在酸性条件(或碱性条件)下的结合,所述条件允许ADA/药物复合物的解离。在一些实施方案中,这些基质具有高装载容量,其改进敏感性,因此允许检测以相对低的浓度存在的ADA和/或药物材料。通常使用的基质的实例包括但不限于碳表面(例如多孔或高结合碳板)、玻璃表面、二氧化硅表面、塑料表面、金属表面、含有金属或化学涂层的表面、膜(例如尼龙、聚砜、二氧化硅)、微珠(例如胶乳、聚苯乙烯,或其他聚合物)、多孔聚合物基质(例如聚丙烯酰胺凝胶、多糖、聚甲基丙烯酸酯)和包含纤维素纤维的基质(例如纤维素海绵、纤维素纸)。在一方面,多孔或高结合碳板是MSD(Meso Scale)高结合板。基质可以是能够感应靶分子的生物传感器芯片、微阵列或芯片上实验室(lab-on-chip)。可应用能够感应与生物传感器的特异性结合的任何种类的生物传感器,包括市售的生物传感器,如由Biacore生产的生物传感器。
如本文使用,通过术语“标记”修饰的实体(例如抗体、抗药物抗体、药物、蛋白、酶、抗体、抗体片段、多域生物治疗剂(例如抗体药物缀合物)或相关物质)包括与另一凭经验可检测的分子或化学实体(例如“可检测的标记物”)缀合的任何实体。适于作为标记物用于标记的实体的化学物质包括但不限于酶、荧光染料、量子点;光染料;发光染料;和放射性核素。
如本文使用的术语“一种或多种”包括至少一种,更适当地,“一种或多种”指一、二、三、四、五、十、二十、五十、一百、五百种等。
本文公开的方式已显示可消除ADA测定中的药物干扰。在实践中,该方法原理可用于减少/消除任何类型的免疫测定中的干扰。该方法原理还可用于任何针对ADA、PK和生物标记物的配体结合测定。本文所述的方法可用于配体结合测定以测试中和抗体(NAb)。配体结合测定可包括与药物靶标结合的药物的竞争性抑制。在PK和生物标记物测定中,可添加过量的抗体用于复合物的形成且沉淀和酸解离后,使用标记的检测抗体进行检测。在全部的情况中,在测定中优化PEG的浓度以平衡敏感性和特异性是重要的。PEG的浓度越高,分子量越低的蛋白将沉淀。因此,为特异性沉淀包含靶分析物的合意的复合物(如抗体药物复合物沉淀),技术人员需要最小化待沉淀的未结合的非特异性蛋白的量(如血清IgM和IgG)。本研究中由于MSD高结合板的碳和多孔结构对其进行利用。基于测定设计原理,其他大容量涂覆表面也将发挥作用。
除非另外表明,本文使用的全部的技术和科学术语与本发明所属领域的技术人员通常理解具有相同的含义。本文描述了用于本发明的方法和材料;还可使用本领域已知的其他适当的方法和材料。所述材料、方法和实例仅是说明性地而非意欲限制。全部出版物、专利申请、专利、序列、数据库登录项和本文提及的其他参考文献在本文通过提述以其整体并入。在冲突的情况中,以本说明书包括定义为主。
本发明的其他特征和优势从下文详述和附图和从权利要求将显而易见。
附图说明
图1是根据本发明的ADA测定的实施方案的示意图。
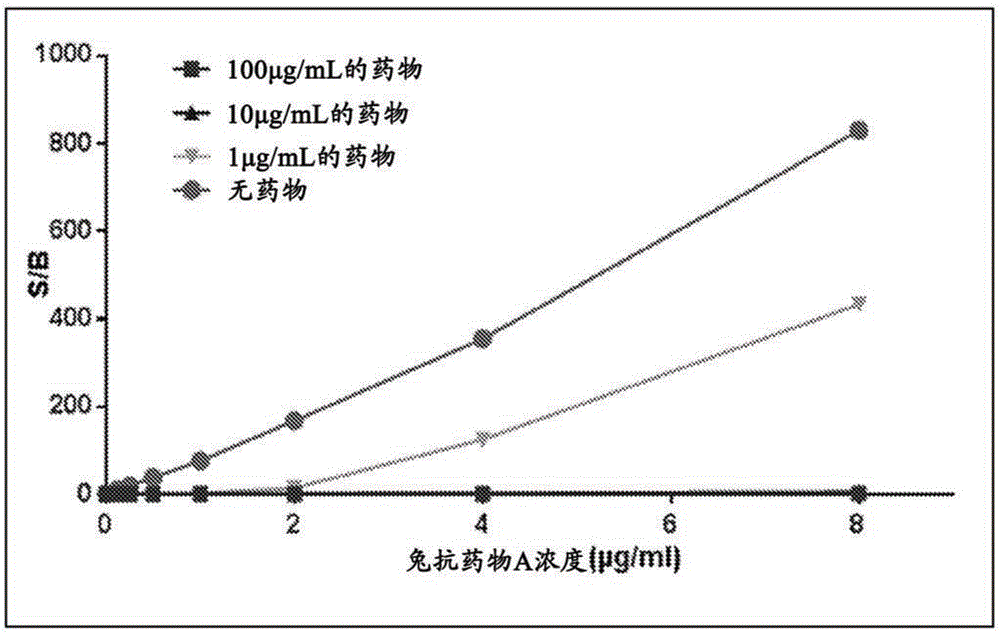
图2A和2B的图描述了检测8μg/mL-125ng/mL水平的亲和纯化的兔抗体与多种浓度的药物A(0、1、10和100μg/mL)的示例性桥接测定形式的结果,其在无酸解离的MSD桥接测定形式中测试。A.针对ADA浓度绘制观察到的S/B以评估与无药物的基线相比具有不同水平的药物的抗体的回收。B.相对基线的百分比回收。ADA:MSD桥接测定:Meso Scale桥接;S/B:信号比背景。
图3A和3B的图描述了检测8μg/mL-125ng/mL水平的亲和纯化的兔抗体与多种浓度的药物A(0、1、10和100μg/mL)的示例性桥接测定形式的结果,其在具有酸解离的MSD桥接测定形式中测试。A.针对ADA浓度绘制观察到的S/B以评估与无药物的基线相比具有不同水平的药物的抗体的回收。B.相对基线的百分比回收。S/B:信号比背景。
图4A和4B的图描述了检测8μg/mL-125ng/mL水平的亲和纯化的兔抗体与多种浓度的药物A(0、1、10和100μg/mL)的示例性桥接测定形式的结果,其在使用聚乙二醇沉淀的MSD桥接测定和本发明的酸解离测定形式(即PandA测定形式)中测试。A.针对ADA浓度绘制观察到的S/B以评估与无药物的基线相比具有不同水平的药物的抗体的回收。B.相对基线的百分比回收。S/B:信号比背景。
图5的图描述了检测无药物的亲和纯化的兔抗药物A抗体的示例性桥接测定形式的结果,其在PEG和酸(PandA)测定形式中使用MSD高结合板的三个不同批次测试。针对ADA浓度绘制观察到的S/B。ADA:抗药物抗体;S/B:信号比背景。
图6的图描述了在收集的正常血清样品(n=32)中检测药物B ADA的示例性桥接测定形式的结果,其在有和无酸解离的MSDB测定形式中评估。正常群体分布(药物B桥接测定)。无处理=无解离,酸处理=具有酸解离。S/B:信号比背景。
图7的图描述了在收集的疾病基线血清样品(n=16)中检测药物B ADA的示例性桥接测定形式的结果,其在有和无酸解离的MSD桥接测定形式,以及PandA测定中评估以确定群体分布。
图8A和8B的图描述了检测4μg/mL-31.3ng/mL水平的亲和纯化的兔抗体与多种浓度的药物B(0、10、50和250μg/mL)的示例性桥接测定形式的结果,其在无酸解离的MSD桥接测定形式中检测。A.针对ADA浓度绘制观察到的S/B以评估与无药物的基线相比具有不同水平的药物的抗体的回收。B.相对基线的百分比回收。S/B:信号比背景。
图9A和9B的图描述了检测4μg/mL-31.3ng/mL水平的亲和纯化的兔抗体与多种浓度的药物B(0、10、50和250μg/mL)的示例性桥接测定形式的结果,其在使用PandA测定形式的MSD桥接测定中测试。A.针对ADA浓度绘制观察到的S/B以评估与无药物的基线相比具有不同水平的药物的抗体的回收。B.相对基线的百分比回收。S/B:信号比背景。
图10A和10B的图描述了检测8μg/mL-125ng/mL水平的亲和纯化的兔抗体与多种浓度的药物C(0、2.5、25和250μg/mL)的示例性桥接测定形式的结果,其在具有酸解离的MSD桥接测定形式中检测。A.针对ADA浓度绘制观察到的S/B以评估与无药物的基线相比具有不同水平的药物的抗体的回收。B.相对基线的百分比回收。S/B:信号比背景。
图11A和11B的图描述了检测8μg/mL-125ng/mL水平的亲和纯化的兔抗体与多种浓度的药物C(0、2.5、25和250μg/mL)的示例性桥接测定形式的结果,其在使用PandA测定形式的MSD桥接测定中评估。A.针对ADA浓度绘制观察到的S/B以评估与无药物的基线相比具有不同水平的药物的抗体的回收。B.相对基线的百分比回收。S/B:信号比背景。
发明详述
本发明的发明人开发了用于定性和/或定量检测来自样品中的ADA的新的方法,所述方法可有效减少并消除由ADA检测中的药物或靶引起的干扰问题。使用PEG沉淀、酸解离、在酸性条件下高容量表面上的涂覆的原理,本文所述的方法允许ADA的特异性检测以及允许使用特异性检测试剂的药物或药物靶标的检测。所述方法可用于更广泛的应用中,以用于减少或消除下述中的干扰:用于ADA、PK/TK的免疫测定和生物标记物(如药物靶标)测定,以及用于检测中和抗体的配体结合测定。
对于具有长半衰期和/或以高剂量或重复剂量施用的药物,如基于抗体的疗法,ADA通常与药物形成循环的免疫复合物,通常使ADA不能检测。例如,循环的药物可干扰ADA和药物靶标的检测,或ADA可干扰药代动力学(“PK”)研究和/或毒代动力学(“TK”)研究中药物水平的检测和/或精确定量。单克隆抗体药物干扰,特别是来自人IgG4药物的干扰由于其较长的半衰期和较高的剂量提出了对于ADA分析的额外挑战。这种干扰的影响特异性针对免疫测定方法和平台且可依赖于在每次各测定中使用的试剂。当药物本身是人源化抗体治疗剂时,耐药免疫原性测定的开发变得更具挑战性。
目前的使用中最广泛使用的方法具有时间、敏感性或精确性的限制,这由与配体测定中结合伴侣相同或相似的干扰因素的存在所导致。这些干扰包括但不限于ADA测定中的药物干扰、PK测定中的ADA和/或药物靶标干扰、药物靶标生物标记物测定中的药物干扰等。通用的ADA方法是桥接测定,其中捕获药物(未标记或生物素标记的)和标记的检测药物间以多价ADA桥接。该形式对内源药物干扰(假阴性ADA)和/或药物靶标干扰(假阳性ADA)易感。作为分析方法开发领域中的主要挑战之一,许多方法已用于减轻该问题如酸或碱解离,第三方结合伴侣竞争性抑制和/或干扰因素的去除、固相提取(SPEAD)、ACE和许多其他方法。样品处理中酸解离配合桥接测定的使用允许药物存在下ADA检测的一定改进,所述测定具有改进的测定敏感性和ADA回收。使用通常允许药物耐性中的一定改进的SPEAD和ACE一般观察到相同的结果,但不能完全维持敏感性和相对精确性。尽管酸(或碱)解离ADA/药物免疫复合物,一旦混合物在测定条件下中和,免疫复合物重新形成。在目前使用的ADA测定中,仅当由于ADA-药物复合物的形成导致ADA的产生超过存在于患者血清中的药物的量时可能检测ADA。这种药物干扰导致产生ADA的患者的患者数目的低估。
药物产品特别是治疗性蛋白的免疫原性是临床和临床前研究中的主要问题,这是由于其可导致潜在的严重副作用、效力的丧失和药物暴露的改变,这使毒性、药代动力学(PK)和药效动力学(PD)数据复杂化。由于具有长半衰期的药物产品如单克隆抗体数目增加,ADA测定中的药物耐性进一步受到关注。为评估药物的整体免疫原性潜力,对ADA的总量特别感兴趣。广泛用于检测血清和血浆中的ADA的多种技术依赖于ADA与其靶药物经由抗原结合位点的特异性结合。例如,桥接测定形式依赖于ADA上抗原结合位点的可用性。由于多种这样的测定形式依赖于ADA上抗原结合位点的可用性,仅可检测到无药物或无部分药物的ADA。由于特定测定材料的信号和时间的紧迫,用于在生物样品中检测ADA的具有改善药物耐性的测定形式是高度需要的。
药物耐性一般定义为仍导致可检测的ADA信号的样品中的游离药物的最大量。样品的酸处理已用于在ADA测定中改进游离药物的耐性。减弱且最终由低pH破坏的抗体-抗原(或药物)结合使从部分或完全药物结合的ADA解离的游离ADA的检测在多种免疫原性测定形式(即桥接测定形式)中成为可能,由此改善药物耐性。然而,如在本文提供的实施例中所证明,酸处理本身不消除ADA测定中的药物耐性。
在一些实施方案中,抗体-抗原(或药物)结合减弱且最终由高pH破坏,使从部分或完全药物结合的ADA解离,随后用碱溶液处理的游离ADA的检测在多种免疫原性测定形式中成为可能。生物药物的结构特征如pI或一些偶共轭键的存在将主宰酸溶液或碱溶液是否更适于破坏ADA/药物结合。为增加药物耐性并提供改善的ADA测定形式,本发明的发明人开发了用于确定具有改善的药物耐性的样品中ADA存在或不存在的方法。预期本文所述方法包括聚乙二醇沉淀步骤和酸解离步骤。
如本文公开,本发明涉及用于确定样品(例如生物样品)中ADA存在或不存在的方法,所述方法包括使样品和结合ADA以形成药物/ADA复合物的过量的药物接触,使所述药物/ADA复合物与聚乙二醇(PEG)接触以形成包含药物/ADA复合物的沉淀,使所述沉淀与溶液接触以解离所述药物/ADA复合物,将所述解离的ADA固定于基质上,和确定所述ADA的存在或量。另一方面,确定步骤包括:使所述固定的ADA与缀合有可检测的标记物的药物接触;和确定所述可检测的标记物存在或量,以由此确定所述样品中ADA存在或不存在。
在一个实施方案中,使沉淀(药物/ADA复合物)与酸溶液接触以解离复合物然后以足以允许全部解离的游离药物和游离ADA涂覆的时间涂覆在大表面(在酸性条件下以保持药物和ADA分开)或基质(例如具有高涂覆容量的高结合碳板)上。当固定至基质表面上时酸性环境防止复合物重新形成。当将酸溶液用于解离复合物时,可将测定称作PandA(PEG和酸)测定。
在另一实施方案中,最终沉淀(药物/ADA复合物)用碱溶液重构以解离复合物然后以足以允许全部解离的游离药物和游离ADA涂覆的时间涂覆至大表面(在碱性条件下以保持药物和ADA分开)或基质(例如具有高涂覆容量的高结合碳板)上。当固定至基质表面上时碱性环境防止复合物重新形成。
酸性或碱性溶液的选择将依赖于药物(例如生物药物)的参数如pI或一些共轭键的存在,且选择将对药物的完整性和结构具有最小影响。
本发明还涉及用于减少药物PK/TK测定中由于生物样品中ADA的存在导致的干扰的方法,所述方法包括使样品与过量的ADA接触以形成药物/ADA复合物,使药物/ADA复合物与聚乙二醇(PEG)接触以由此形成包含药物/ADA复合物的沉淀,使所述沉淀与酸溶液接触由此解离药物/ADA复合物,将解离的游离药物和游离ADA固定至表面和/或基质上,并使用针对药物的特定检测试剂实施药物PK/TK测定以由此减少来自ADA的干扰。药物测定可以是例如药物定量测定、药物PK/TK测定或药物效力测定。可将相似的原理应用于定量药物靶标的方法作为在药物干扰存在下用于药物安全性和效力(PD)的生物标记物测定。将过量的药物添加至样品以形成药物靶标/药物复合物。使用PEG沉淀和酸解离(或碱解离)步骤然后使用特定药物靶标检测试剂。
在一些方面,本公开内容提供用于确定针对药物的ADA的存在或不存在的方法。当引入人或其他动物的身体时,如本文使用的术语“药物”或“药物材料”指具有药用、性能增强和/或兴奋作用的化学物质。例如,药物可以是有机的或无机的小分子化合物或生物治疗剂(例如抗体(例如药物抗体)或其片段,多重域生物治疗剂、核酸、肽、多肽、肽模拟物、糖或脂质),只要药物是免疫原性的且能够诱发免疫响应。术语“药物抗体”指可施用至个体用于治疗疾病的抗体,且如本文使用的这些抗体与ADA不同。药物抗体的非限制性实例包括例如选自下组的抗体:muromomab-CD3、阿昔单抗、利妥昔单抗、达克珠单抗、巴利昔单抗、帕利珠单抗、英利昔单抗、曲妥珠单抗、依那西普、吉姆单抗、fresolimumab、阿仑单抗、ibritomomab、阿达木单抗、阿来塞普、奥马珠单抗、tofacitinib、托西莫单抗、依法利珠单抗、西妥昔单抗、贝伐珠单抗、那他珠单抗、兰尼单抗、帕尼单抗、依库珠单抗、美泊利单抗、necitumumab、blinatumomab、nivolumab、dinutuximab、secukinumab、evolocumab、pembrolizumab、ramucirumab、vedoluzumab、siltuximab、opinutuzumab、adotrastuzumab、emtansine、raxibacumab、帕妥珠单抗、brentuximab、贝利单抗、易普利姆玛、地诺单抗、托珠单抗、ofatumumab、canakinumab、戈利木单抗、ustekinumab、catumaxomab和赛妥珠单抗。
本文公开的方法可包括聚乙二醇(“PEG”)介导的沉淀步骤,所述步骤包括使样品与PEG化合物接触。所述PEG化合物可以是具有1,000-40,000道尔顿的分子量的PEG化合物。例如,所述PEG化合物包括至少一种选自下组的PEG:PEG1000、PEG1450、PEG3000、PEG6000、PEG8000、PEG10000、PEG14000、PEG15000、PEG20000、PEG250000、PEG30000、PEG35000和PEG40000。
选自PEG的具体浓度以最大平衡特异性和选择性。与样品接触的PEG的量可对应如下浓度:约0.1%-约10.0%、约0.2%-约7.0%、约0.5%-约6.0%、约0.5%-约5.0%、约1.5%-约5.5%、约2.0%-约5.0%、约3.0%-约4.5%、约3.5%-约4.0%、约1.0%-约2.5%、约1.2%-约1.5%或约0.1%、0.2%、0.5%、1.0%、1.2%、1.5%、2.0%、2.5%、3.0%、3.5%、4.0%、4.5%、5.0%、5.5%、6.0%、6.5%、7.0%、7.5%、8.0%、8.5%、9.0%、9.5%或约10.0%PEG。
所述方法可包括酸解离步骤,所述步骤包括使沉淀与酸接触。酸可以是或包括有机酸。可替换地或此外,酸可以是或包括无机酸。在解离步骤中使用的酸可包括有机酸和无机酸的混合物。有机酸的非限制性实例包括例如柠檬酸、异柠檬酸、谷氨酸、乙酸、乳酸、甲酸、草酸、尿酸、三氟乙酸、苯磺酸、氨基甲磺酸、樟脑-10-磺酸、氯乙酸、溴乙酸、碘乙酸、丙酸、丁酸、甘油酸、琥珀酸、苹果酸、天冬氨酸及其组合。无机酸的非限制性实例包括例如盐酸、硝酸、磷酸、硫酸、硼酸、氢氟酸、氢溴酸及其混合物。
酸的量可对应下述浓度:约0.01M至约10M、约0.1M至约5M、约0.1M至约2M、约0.2M至约1M或约0.25M至约0.75M的酸或酸的混合物。在一些实例中,酸的量对应大于或等于下述的浓度:约0.01M、0.05M、0.1M、0.2M、0.3M、0.4M、0.5M、0.6M、0.7M、0.8M、0.9M、1M、2M、3M、4M、5M、6M、7M、8M、9M或10M的酸或酸的混合物。酸的pH可以是例如约0.1、0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0、5.5、6.0或6.5。
所述方法可包括碱解离步骤,所述步骤包括使沉淀与碱接触。碱可以是或包括有机碱。可替换地或此外,碱可以是或包括无机碱。在解离步骤中使用的碱可以包括有机碱和无机碱的混合物。碱的非限制性实例包括例如尿素、氢氧化钠、氢氧化铷、氢氧化铯、氢氧化钙、氢氧化锶、氢氧化钡、氢氧化锌、氢氧化锂、丙酮、甲胺和氨,及其混合物。
当将碱溶液用于破坏ADA/药物相互作用时,碱的量可对应下述浓度:约0.01M至约5M、约0.1M至约5M、约0.1M至约1M、约0.2M至约1M或约0.25M至约0.75M的碱或碱的混合物。在一些实例中,碱的量对应大于或等于下述浓度:约0.01M、0.05M、0.1M、0.2M、0.3M、0.4M、0.5M、0.6M、0.7M、0.8M、0.9M、1M、2M、3M、4M、5M、6M、7M、8M、9M或10M的碱或碱的混合物。碱的pH可以是例如约8.0、8.5、9.0、9.5、10.0、10.5、11.0、11.5、12.0、12.5和13.0。
在一些实施方案中,使样品与酸或碱以足以解离预先形成的药物/ADA复合物的时间量接触。在一些实例中,使样品与酸或碱以下述时间段接触(例如温育):约0.1小时至约24小时,例如约0.2小时至约16小时、约0.5小时至约10小时、约0.5小时至约5小时或约0.5小时至约2小时。在另外的实例中,使样品与酸或碱以大于或等于下述的时间段接触(例如温育):约0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、1.5、2、2.5、3、3.5、4、4.5、5、6、7、8、9或10小时。所述样品可与酸或碱以在任何一般与所述方法兼容的温度接触,例如4℃、室温(RT)或37℃。RT可以是例如22℃-26℃,例如23℃、24℃或25℃。
所述方法可包括将解离的ADA和/或药物固定至表面和/或基质上。基质可包括碳表面、玻璃表面、二氧化硅表面、金属表面、用聚合材料涂覆的表面、含有金属或化学涂层的表面、膜、微珠、多孔聚合物基质。基质可包括多孔碳表面。在示例性的实施方案中,基质是具有大表面和高涂覆容量的高结合碳板。
所述方法可进一步包括确定样品中ADA的存在或量,或药物的存在或量。因此,本文提供了抗体、抗药物抗体或用可检测的标记物标记的药物。用于本发明任何方法的可检测的标记物的非限制性实例包括半抗原、酶、酶底物、酶抑制剂、荧光团、发色团、发光标记物、放射性同位素(包括放射性核素)和结合对的成员。可检测标记物的强度可使用本领域的技术人员已知的仪器和设备测量,包括例如可携带或台式荧光计(例如手持式荧光计)或可携带或台式比色计(例如手持式比色计)。
在一些实施方案中,可检测的标记物是电化学发光标记物,包括例如标记物。
可检测的标记物可以是结合对的特定成员(第一成员或第二成员)。用于在本文提供的方法中使用的结合对包括例如生物素/链霉亲和素、生物素/抗生物素蛋白、生物素/中性抗生物素蛋白、生物素/凯普亲和素、表位/抗体、蛋白A/免疫球蛋白、蛋白G/免疫球蛋白、蛋白L/免疫球蛋白、GST/谷胱甘肽、His-标签/镍、抗原/抗体、FLAG/M1抗体、麦芽糖结合蛋白/麦芽糖、钙调蛋白结合蛋白/钙调蛋白、酶-酶底物和受体-配体结合对。在一些实施方案中,GlcNac结合蛋白缀合至结合对的第一成员(例如生物素、抗生物素蛋白、中性抗生物素蛋白、凯普亲和素、抗体、抗原、蛋白A、蛋白G、蛋白L、GST、His-标签、FLAG、MBP、钙调蛋白结合蛋白、酶、受体或配体)。
如本文使用的术语“荧光标记物”和“荧光团”可互换使用且指当物质通过不同的波长激发(激发波长)发光时,发射电磁能量如一定波长的光(发射波长)的任何物质,且意在涵盖能够与在样品中的感兴趣的分析物相互作用或特异性反应以提供一种或多种光信号的化学物质或生化分子或其片段。
用于在本文提供的方法中的代表性荧光团包括例如绿色荧光蛋白、蓝色荧光蛋白、红色荧光蛋白、荧光素、荧光素5-异硫氰酸酯(FITC)、花青染料(Cy3、Cy3.5、Cy5、Cy5.5、Cy7)、Bodipy染料(Invitrogen)和/或Alexa Fluor染料(Invitrogen)、丹酰、丹磺酰氯(DNS-Cl)、5-(碘乙酰胺)荧光素(5-IAF、6-丙烯酰-2-二甲氨基萘(Acrylodan)、7-硝基苯并-2-氧杂-1,3,-二唑-4-基氯化物(NBD-Cl)、溴化乙锭、萤光黄、罗丹明染料(5-羧基罗丹明6G氢氯化物、丽丝胺罗丹明B磺酰氯、罗丹明-B-异硫氰酸酯(RITC(罗丹明-B-异硫氰酸酯)、罗丹明800);四甲基罗丹明5-(和6-)异硫氰酸酯(TRITC))、Texas RedTM,磺酰氯、萘磺酸,包括但不限于1-苯胺萘-8-磺酸(ANS)和6-(对-甲苯胺基)萘-e-2-磺酸(TNS)、氨茴酰脂肪酸、DPH、十八碳四烯酸、TMA-DPH、芴基脂肪酸、荧光素-磷脂酰乙醇胺、Texas红-磷脂酰乙醇胺、芘基-磷脂酰胆碱、芴基-磷脂酰胆碱、部花青540、苯乙烯基萘、3,3'二丙基硫代二碳青色素(diS-C3-(5))、4-(对二戊基氨基苯乙烯基)-l-甲基吡啶盐(di-5-ASP)、Cy-3碘乙酰胺、Cy-5-N-羟基琥珀酰亚胺、Cy-7-异硫氰酸酯、IR-125、噻唑橙、天青B、尼罗蓝、铝酞菁、噁嗪1,4',6-二脒-2-苯基吲哚.(DAPI)、Hoechst 33342、TOTO、吖啶橙、乙锭同型二聚体、N(乙氧羰基甲基)-6-甲氧基喹啉(MQAE)、呋喃-2、钙绿、羧基SNARF-6、BAPTA、香豆素、Phytofiuors、Coronene(晕苯)和金属配体复合物。
可用于在本文提供的方法中使用的半抗原包括例如地高辛和生物素。
在本文提供的方法中使用的酶包括例如碱性磷酸酶(AP)、β-半乳糖苷酶、辣根过氧化物酶(HRP)、大豆过氧化物酶(SBP)、尿素酶、β-内酰胺酶和葡萄糖氧化酶。
一些方面,本文提供适用于确定生物样品中ADA的存在或不存在的试剂盒。所述试剂盒可包括说明和在容器中用于使药物/ADA复合物与聚乙二醇(PEG)接触以形成包含药物/ADA复合物的沉淀的试剂和用于使沉淀与酸溶液接触以解离药物/ADA复合物的试剂;和适于将解离的ADA或药物固定用于进一步分析的基质。
实施例
本发明进一步以下述实施例进行描述,所述实施例不限制权利要求中所述的本发明的保护范围。
下文的实施例描述了新型ADA测定方法,其中尽管存在高水平的抗体治疗剂,抗体的完全回收以定量极限获得。具体地,提供了三例研究以证明对于单克隆抗体治疗剂(药物A、B和C)ADA测定中药物干扰的消除。
1.1测定形式
为改善常规免疫原性测定的药物耐性(或消除药物干扰),发明人力求开发新的用于确定样品(例如生物样品)中ADA的存在或量的方法。发明人已成功地开发了新的方法,其中尽管存在高水平的药物(抗体治疗剂),抗药物抗体的完全回收以定量极限获得。所述方法示例性实施方案的示意图在图1中显示。
如图1中所示,将过量的药物材料添加至样品(例如生物样品)以允许药物/ADA复合物形成。初始温育后,将PEG添加至每个样品并温育以允许复合物沉淀。系列洗涤(例如一次或多次)后,用酸溶液重构沉淀以解离药物/ADA复合物。将解离的药物/ADA复合物涂覆在基质上(即涂覆在高结合MSD板的孔上)。温育后,封闭基质然后使用以可检测的标记物(允许通过ECL(“电化学发光”)或“增强的化学发光”的ADA检测)标记的药物实施检测。
一方面,图1中所述的方法包括将过量的药物材料添加至样品以饱和游离的抗体因此形成药物/ADA复合物。复合物随后使用PEG沉淀。将PEG以优化的浓度添加至样品以在维持特异性的同时实现合意的敏感性。系列洗涤后,用酸溶液重构最终沉淀并涂覆在高结合碳板上。酸性环境在复合物吸附至MSD高结合板的多孔碳表面上的同时防止其重新形成。总ADA水平的检测随后使用标记物缀合的药物实施,随后在Meso ScaleSector 2400电化学发光读取器上读取。
1.2实验材料
治疗性单克隆抗体、药物和亲和纯化的兔抗药物由Genzyme,a Sanofi Company(Framingham,MA)开发。来自健康个体的天然人血清汇集物购自Bioreclamation Inc.。疾病基线血清样品从参与产品相关的临床试验的未经治疗的受试者获得。高结合96孔Meso Scale板、Sulfo Tag、Read Buffer T,Sector 2400读取器获得自Meso Scale(“MSD”)。PEG8000获得自TekNova。冰乙酸由J.T Baker提供。吐温20和脱脂干奶粉Sigma从Aldrich获得。ELx405板洗涤剂由Biotek提供。板洗涤缓冲剂来自PerkinElmer。透明聚丙烯96孔微孔板购自Corning。牛血清白蛋白获得自Seracare。硼酸盐可由Sigma Aldrich,目录号B0394获得。
药物A是人源化IgG1消耗抗体,其与自身免疫性疾病的淋巴细胞表面靶标结合。药物B是全长的人IgG4,其中和在纤维化适应症的靶组织中结合于其细胞表面受体的可溶性细胞因子。药物C是人源化IgG4,其识别细胞表面粘附分子并阻断其与可溶性配体的结合。
1.3无酸解离步骤的桥接免疫测定
MSD桥接测定形式需要药物以生物素标记和以标记。根据MSD测定形式,生物素化的药物将作为捕获分子发挥作用且标记的药物在桥接测定中将为报道物。
样品最初在测定缓冲剂中1:10稀释((300mM乙酸,2%BSA)。将样品添加至聚丙烯板的重复的孔中并添加在测定缓冲剂中的包含等摩尔浓度的生物素-药物和Sulfo-TAG-药物的溶液。随后在22-26℃、450rpm的摇床中温育平板2小时。链霉亲和素涂覆的MSD平板用测定缓冲剂封闭最少1小时。温育后,洗涤MSD板3次并将50μL温育的混合物从聚丙烯板转至MSD板并在22-26℃的摇床上温育2小时。温育后,洗涤平板并添加包含三丙胺的MSD读取缓冲剂T的溶液,并在MSD Sector成像仪2400上读取平板。
在仪器内,施加电压。在三丙胺存在下,在电化学发光(ECL)反应中沉淀。抗体桥接与链霉亲和素表面结合的Sulfo-TAG-药物和生物素-药物将导致ECL信号。最终温育后,用在PBS中的0.05%吐温洗涤平板。随后添加读取缓冲剂T 2x并在Sector PR2400读取平板。电化学发光信号与每个样品中的抗药物抗体是成比例的。样品的结果通过用来自个体样品的平均ECL信号除以阴性对照的平均ECL信号转化为信号比背景的比率(S/B)。
1.4具有酸解离步骤的桥接免疫测定
样品最初在测定缓冲剂(300mM乙酸,2%BSA)中1:10稀释,在22-26℃温育45分钟。随后将样品添加至聚丙烯平板的重复的孔中,并以有效中和pH至7.0的比率添加在测定缓冲剂中的包含等摩尔浓度的生物素-药物和Sulfo-TAG-药物以及Tris HCL的溶液。随后在22-26℃的摇床以450rpm温育平板2小时。将链霉亲和素涂覆的MSD平板用测定缓冲剂封闭最少1小时。温育后,洗涤MSD平板3次随后将温育的混合物从聚丙烯平板转移至MSD平板并在22-26℃摇床上温育2小时。温育后,洗涤平板并添加包含三丙胺的MSD读取缓冲剂T的溶液并在MSD Sector成像仪2400上读取平板。
在仪器内,施加电压。在三丙胺存在下,在电化学发光(ECL)反应中沉淀。抗体桥接与链霉亲和素表面结合的Sulfo-TAG-药物和生物素-药物将导致ECL信号。最终温育后,用在PBS中的0.05%吐温洗涤平板。随后添加读取缓冲剂T 2x并在Sector PR2400读取平板。电化学发光信号与每个样品中的抗药物抗体是成比例的。样品的结果通过用来自个体样品的平均ECL信号除以阴性对照的平均ECL信号转化为信号比背景的比率(S/B)。
1.5PEG和酸(PandA)步骤1
样品最初在包含过量药物(10-50μg/mL)的测定缓冲剂(300mM乙酸,2%BSA)中1/5稀释并在聚丙烯平板中于37℃以450rpm温育1小时以允许药物和样品中任何残留的游离抗体之间的复合。随后添加pH 8.0硼酸盐中的3%PEG至每个样品并在2-8℃温育过夜。每个样品中PEG缓冲液的最终浓度为1.5%。
下一天,将平板在4000rpm离心20分钟以将复合物沉淀成丸。所述丸随后用硼酸盐pH 8.0中的1.5%PEG重悬并在4000rpm第二次离心20分钟。将洗涤循环重复三次。最终浓缩后,每个样品在100μl的300mM乙酸中悬浮并进一步1/10稀释(20μl样品+180μl乙酸),使最终样品稀释为1/50。随后通过添加25μl的一式两份至MSD高结合平板的孔涂覆稀释的样品,并在24℃以450rpm振荡1小时。温育后,用1x平板洗涤缓冲剂洗涤平板,并在24℃振荡用PBS中的3%奶封闭1小时,其后,洗涤平板并将100ng/ml的Sulfo-TAG-药物添加至样品并在24℃振荡中温育1小时。最终温育后,用在PBS中的0.05%吐温洗涤平板。随后添加读取缓冲剂T 2x并在Sector PR2400上读取平板。电化学发光信号与每个样品中的抗药物抗体成比例。
在一些实施方案中,初始酸添加后的温育步骤可在23℃、25℃、27℃、30℃、32℃、35℃、37℃、39℃或更高实施。
在一些实施方案中,最终沉淀步骤后,每个样品可进一步稀释至例如1:5、1:10、1:20、1:25、1:30、1:40、1:50或1:60、1:80、1:100或1:200的最终样品稀释度。一般的最终样品稀释度可为<1:100,其为食品和药物监督管理局(FDA)设定的最低所需稀释度(MRD)。
1.5PEG和酸(PandA)步骤2
样品最初在包含过量药物(10-50μg/mL)的测定缓冲剂(300mM乙酸,2%BSA)中1/5稀释并在聚丙烯平板中于37℃以450rpm温育1小时以允许药物和样品中任何残留的游离抗体之间的复合。随后以1:1的PEG:样品比例将PEG溶液添加至每个样品并在2-8℃温育过夜。对于每个产品优化PEG的最终浓度以使特定复合物最佳沉淀,并实现合意的特异性和敏感性同时最小化未复合的分子如非特异性IgG的作用。添加至样品的PEG浓度为3%-6%(以1:1的比率添加至稀释样品的药物A为3%且药物B和C为6%)。
下一天,将平板在4000rpm离心30分钟以将复合物沉淀成丸。所述丸随后用PBS中的PEG重悬并在4000rpm第二次离心20分钟。将洗涤循环额外重复一次。最终离心后,每个样品在300mM乙酸中重悬并稀释以实现对于特定测定合意的最终MRD(药物A和C为1/50且药物B为1/25)。随后将样品涂覆在MSD高结合平板的重复的孔中。随后在24℃以450rpm振荡温育平板1小时。温育后,用1x平板洗涤缓冲剂洗涤平板并在24℃振荡用PBS中的3%奶封闭1小时。随后,洗涤平板并将含有Sulfo-TAG-药物的溶液添加至样品并在24℃振荡温育1小时。最终温育后,用1x平板洗涤缓冲剂洗涤平板。随后添加读取缓冲剂T 2x并在Sector PR2400上读取平板。电化学发光信号与每个样品中的抗药物抗体成比例。
在一些实施方案中,初始酸添加后的温育步骤可在23℃、25℃、27℃、30℃、32℃、35℃、37℃、39℃或更高实施。
在一些实施方案中,最终沉淀步骤后,每个样品可进一步稀释至例如1:5、1:10、1:20、1:25、1:30、1:40、1:50或1:60、1:80、1:100或1:200的最终样品稀释度。一般的最终样品稀释度可为<1:100,其为食品和药物监督管理局(FDA)设定的最低要求稀释度(MRD)。
2.1药物A
敏感性药物耐性评估
对于药物A,比较PandA方法和具有或不具有酸解离的常规MSD桥接测定以改善药物耐性。测定敏感性和药物耐性使用8μg/mL-125ng/mL浓度的亲和纯化的兔抗药物确定,其在MSD桥接测定形式中,具有或不具有多种浓度(0、0.1、10和100μg/mL)药物(药物A)、具有(图2A-B)和不具有(图3A-B)酸解离。包含ADA和药物的样品在汇集的正常人血清中制备并在37℃温育至少1小时允许药物/ADA复合物在测定前形成。
通过评价40个正常人血清样品、计算样品的均值和标准偏差并计算1.645倍标准偏差的第95百分位的因子并将其添加至均值来确定切割点(cut point),如通常所建议的。对于药物A,将新的方法与具有或不具有酸解离的常规桥接测定进行比较用于药物耐性。
图2是比较无酸解离的MSD桥接测定的数据总结。结果指示药物不存在下对于ADA检测的强剂量响应,且用1μg/mL的药物看到抑制(图2A)。图2B指示抗体检测的低回收率,在所检测的1μg/mL最低药物浓度在125ng/mL的ADA为约10%。(图2B)药物不存在下,用1μg/mL的药物,测定敏感性从15ng/mL减少至342ng/mL。100μg/mL的药物存在下,敏感性减少至5143ng/mL或5.1μg/mL。
图3是来自具有酸解离的MSD桥接测定形式的数据总结。药物不存在下对于ADA检测,结果指示对无酸的桥接测定的相似的剂量响应,且用1μg/mL的药物看到抑制。(图3A)1μg/mL药物的百分比回收率仍是可接受的,但在低于药物Cmax的10μg/mL药物,在125ng/mL的ADA回收率减少至35%。(图3B)测定检测敏感性对于1和10μg/mL的药物维持在约15ng/mL,而100μg/mL药物存在下,发现为262ng/mL。尽管测定的敏感性满足该方法中提出的250-500ng/mL的准则,该发现对该产物和抗体组合是特异性的且对于其他产物或以不同抗体对照是不可接受的。
图4A和4B是来自PandA(步骤2)沉淀形式的数据的总结。结果指示药物不存在下的可接受的剂量响应,且由于在样品中存在药物而未看到显著抑制。(图4A)当与不具有药物作为参照的样品结果进行比较时,百分比回收率保持为可接受的大多为80-120%而无论样品中存在的药物的量。(图4B)相似地,尽管药物以100μg/mL存在(比药物A的预期Cmax高3-4倍)但测定检测敏感性维持在9-14ng/mL。
表1是每种测试的方法中,在测试的药物A的多种浓度对于ADA检测的测定敏感性的总结。测定敏感性浓度通过从图2A、3A和4A所示的每个抗体曲线回拟合S/B切割点而获得。ADA:抗药物抗体;S/B:信号比背景。
表1.
如表1所示,尽管存在100μg/mL的药物,PandA方法不仅改进药物耐性还将测定敏感性维持在9-14ng/mL。抗体检测回收率保持在大多80-120%而无论存在于样品中药物的量,其优于具有酸解离的桥接免疫测定。
效价结果比较
PandA方法还可用于精确报告药物存在下的抗体效价。为确定终点效价是否在仅包含抗体的样品(0μg/ml药物)和包含等量抗体和高药物浓度(100μg/mL药物)的其他样品之间相关,在其中两种不同滴定方案后实施滴定稀释的方法中分析测试样品。第一方案在PEG沉淀前并入滴定且第二方案在涂覆于高结合平板前并入滴定。终点效价在相同抗体水平的无药物和包含药物的样品之间以及在基于测定效价切割点值(1.2的S/B)的滴定方案之间相同。终点效价数据总结与表2。
表2.
3.1.MSD高结合平板(批对批(lot to lot))和整体精确性
为确定MSD高结合平板是否有助于测定变化性,样品的涂覆在三个不同批次上实施。绘制数据并使用ANOVA对等价性进行分析。
为确定MSD高结合平板是否有助于测定变化性,样品的涂覆在三个不同批次上实施。样品包含了具有多种浓度的药物的亲和纯化的兔抗体。如图5所示,针对平板的三个批次的每一个的ADA浓度绘制观察到的S/B。平板批次间的精确性发现是可接受的,其中CV小于20%(表3)且ANOVA在三个批次间未显示显著差异,其中p值为0.1776。
4.1药物B
具有酸解离的MSD桥接测定中的靶干扰
对于药物B,在具有酸解离的MSD桥接测定中看到特定的挑战,这是由于药物B的靶标在低pH从单体变为二聚体引起假阳性结果。在具有酸解离的MSD桥接测定中,在100%正常血清样品和疾病基线样品中看到二聚化效应。该现象与Dai等[Dai S,Schantz A,Clements-Egan A,Cannon M,Shankar G:Development of a method that eliminates false-positive results due to nerve growth factor interference in the assessment of fulranumab immunogenicity.AAPS J.16(3),464-477(2014)]所报导的相似,其中发现了抗药物抗体(ADA)在阶段1研究中的明显高的发生率是同二聚体药物靶标检测的结果,这是由于其桥接药物分子的能力。Dai等发现用于缓解药物干扰的样品的基于酸解离的预处理显著增加了药物靶标干扰。
为证明测定中由于酸解离和假阳性结果导致的药物靶标的内源性靶标二聚化效应,正常的(n=32)和基线疾病(n=16)血清样品在具有或不具有酸解离的桥接测定中分析以突出测定中由于酸解离和假阳性结果导致的内源性靶标二聚化的效应。
图6是在具有或不具有酸解离的MSD桥接测定中检测的正常血清样品的代表。在无酸处理组中观察到测定背景或其附近的正常分布(左侧群体),而当将酸解离应用至桥接测定时以大于20的S/B观察到一些阳性结果。
图7是当在无或有酸解离的MSD桥接测定以及PEG和酸(PandA)方法中分析时,疾病基线血清样品的代表。在无酸处理的MSD桥接和PandA方法之间结果是可比较的,而酸处理导致大量测试样品的更高S/B水平,指示由于在低pH的二聚化效应所致的来自药物靶标的干扰。
如图6和7所示,对于药物B,具有酸解离的桥接测定在正常或疾病群体中是不可行的,这是由于在较低pH药物靶标的二聚化引起全部样品的假阳性结果,其中结果与内源性靶标的量成比例。由于该原因,药物B的测定敏感性和药物耐性仅在PandA方法和现有的无酸解离的MSD桥接测定之间是可比较的。
图8A-B和9A-B是比较无酸解离的桥接测定形式与PEG和酸方法(PandA)的药物B的数据总结。
图8A中,MSD桥接测定形式导致药物不存在下ADA检测的可接受的剂量响应,且用10μg/mL看到完全抑制且敏感性从不存在药物下的47ng/mL减少至具有10μg/mL药物的2μg/mL抗体。
对于药物B,无酸解离的桥接测定敏感性在50ng/mL验证,其中药物耐性不良。250ng/mL的药物水平抑制250ng/mL的抗-药物B抗体的检测且1μg/mL的药物水平抑制500ng/mL的抗体检测。
图9A-B是来自PandA沉淀形式的数据的总结。结果指示药物不存在下可接受的剂量响应且未看到由于样品中药物存在所致的抑制。当与无药物作为参照的样品结果相比时,百分比回收率保持为可接受的大多为80-120%而无论样品中存在的药物量。测定检测敏感性维持在39-63ng/mL,尽管药物以高于预期Cmax的250μg/mL存在。
5.1药物C
敏感性和药物耐性评估
对于药物C,在新方法和其中不能实现预期药物耐性的现有的具有酸解离的MSD桥接测定之间,比较了测定敏感性和药物耐性。
图10A-B和11A-B是比较具有酸解离的桥接测定形式与PEG和酸方法(PandA)的药物C的数据总结。
在图10A-B中,MSD桥接测定形式导致药物不存在下可接受的敏感性,但尽管桥接免疫测定中进行酸处理,以少至2.5μg/mL的药物可见抑制。25μg/mL存在下测定的敏感性从227ng/mL变为2788ng/mL且在250μg/mL的药物(在一些临床样品中预期的水平)存在下抗体完全是不可检测的
图11A-B显示PandA结果优于具有酸解离的桥接免疫测定,其中即使存在250μg/mL的药物,抗体检测以完全的回收维持。当与无药物作为参照的样品结果相比时,观察到百分比回收率大多为80-120%无论样品中存在的药物量。尽管药物以250μg/mL(高于临床样品中所预期的)存在,测定检测敏感性维持在129-175ng/mL。
上文实施例描述了三种人源化单克隆抗体A-C(IgG1和2种IgG4药物)的病例研究。
相比MSD桥接测定中通常使用的测定解离方法,三种药物特异性PandAADA测定导致包含超过患者中所预期的药物水平的样品中ADA的完全回收。该突破性的新方法显示超越现有方法的显著改进。事实上,在全部三种情况中药物干扰或ADA的低检测完全消除,且该测定原理可不仅用于ADA测定还可用于干扰因素存在下的PK和生物标记物(药物靶标)分析。
本文所述的PandA ADA测定方法显示可有效改进包含超过临床相关的干扰水平的样品中ADA的检测和回收。所述方法还报告一致的抗体效价,而无论药物的存在量。该方法显示优于基于具有酸解离的桥接测定的常规方案,其在超过预期临床Cmax水平的药物水平维持ADA检测敏感性且无显著抑制。
PandA方法还可应用至PK测定,其中ADA或药物靶标是干扰性的。简单地说,添加过量的抗体(抗独特型)或靶标以形成复合物且将使用特异性针对药物的标记的抗独特型进行检测。此外,所述方法可应用于具有潜在药物干扰的药物靶标生物标记物的测定。总之,发明人已描述了复合物PEG沉淀的新应用,其分解药物并可能靶向ADA免疫测定中的干扰。
多种不同的方法和平台已以有限的成功用于解决检测ADA的免疫测定中的循环药物干扰。本文公开了采用PEG和酸(PandA)的新方法,其消除ADA免疫测定中药物和可能的靶标干扰。新方法显示在高药物浓度的药物干扰的完全消除并证明了相对于基于无或甚至具有酸解离的MSD桥接测定的常规方案维持测定敏感性。通过采用下列测定组分,PandA测定已证明其在药物和/或药物靶标存在下选择性和特异性地检测ADA的意欲用途:添加过滤药物材料以形成药物/ADA复合物;使用聚乙二醇沉淀以得到总ADA;酸解离并在酸性溶液中在具有高容量的高结合碳板上涂覆重构的沉淀物以允许结合解离的ADA;并用ECL输出特异性地检测总ADA水平缀合药物。
此外,该方法还报导一致的抗体效价,而无论存在于样品中的药物量,其显示测定精确性。还报导的是,方法有效解决了由于在具有酸解离的MSD桥接免疫测定中的靶二聚化引起假阳性结果的靶干扰。
总之,所述方法优于具有酸解离的常规桥接方法,如通过上述报告的三项主要研究证据所证明的,其中用样品中的高药物量实现了样品中ADA的完全回收和检测。根据目前的监管预期和测试的临床样品成功验证了该方法。
本文所述的PandA方法已显示在过量药物存在下对于ADA检测的显著改善。基于原理其具有广泛应用:(1)饱和游离的分析物以形成复合物中全结合的分析物和(2)沉淀复合物(3)酸解离以游离分析物而无中和,从而在酸性条件下将游离的分析物涂覆至大涂覆表面上来固定游离的分析物时减少分析物的再结合,和(4)使用特定的试剂检测游离的分析物。在上述实施例中,发明人已提供了三种免疫原性病例研究以证明该新技术的效用。
其他实施方案
应该理解的是尽管本发明已结合其发明详述进行了描述,上文的描述意欲说明而非限制本发明的范围,所述范围通过所附的权利要求书定义。其他方面、优势和修饰都在下述权利要求书的范围内。
- 还没有人留言评论。精彩留言会获得点赞!