本发明属于医药技术领域,具体涉及还原型谷胱甘肽降解产物F的分离纯化方法,即(2S)-2-氨基-4-[1-(羧甲基)氨甲酰基-(2R)-2-亚磺酰乙基氨甲酰基]丁酸的分离纯化方法,以及其在还原型谷胱甘肽有关物质检测中作为杂质对照品的用途。
背景技术:
还原型谷胱甘肽为自然界广泛存在的含有巯基(-SH)的三肽,化学名为N-(N-L-γ-谷氨酰基-L-半胱氨酰基)甘氨酸,结构式如下:
还原型谷胱甘肽通过生物发酵技术所得,属于含有巯基(-SH)的三肽化合物,其在自然界中亦广泛存在,主要存在于细胞质中,在多种细胞生化功能中起作用。还原型谷胱甘肽是甘油醛磷酸脱氢酶的辅基,又是乙二醛酶及磷酸丙糖脱氢酶的辅酶,参与体内三羧酸循环及糖代谢。它能激活体内SH酶等,促进碳水化合物、脂肪及蛋白质的代谢。还原型谷胱甘肽还可通过巯基与体内的自由基结合,促进易代谢的低毒化合物的形成,因此对部分外源性毒性物质具有减毒作用。
还原型谷胱甘肽的水溶性好,可口服,也可肌肉注射或静脉滴注给药,因其具有疗效确切、副作用小的特点而被国内外广泛应用于临床。近年来,还原型谷胱甘肽制剂有关的不良反应亦有报道,主要不良反应表现为:偶见脸色苍白,血压下降,脉搏异常等类过敏症状,应停药;偶见皮疹等过敏症状,应停药;偶有食欲不振、恶心、呕吐、胃痛等消化道症状,注射局部轻度疼痛,停药后消失。还原型谷胱甘肽引起不良反应的机制尚不明确,医护人员和药品生产经营企业应关注还原型谷胱甘肽相关制剂的不良反应,探究其不良反应发生机制,提高药物质量,保证用药安全。
药品中的杂质按化学类别和性质一般分为三类:有机杂质、无机杂质及残留溶剂。有机杂质包括工艺中引入的杂质和降解产物,可能是已知的或未知的,挥发性的或不挥发性的,这类杂质的化学结构与活性成分的分子式类似或具渊源关系,故通常又可称之为有关物质,亦是药物不良反应产生的一个重要活性物质。还原型谷胱甘肽结构中含有还原性较强的巯基(-SH),在生产、储备以及应用过程中与空气中氧气接触,易被氧化产生杂质,在还原型谷胱甘肽原料的生物发酵及制剂的制备过程中,也可能会产生其他降解杂质;这些杂质的引入,对产品质量有较大的影响,尤其是注射用还原型谷胱甘肽(注射剂),由于其直接输注或注射入血液,对机体影响更直接,风险更大,重视还原型谷胱甘肽制剂的杂质(任何影响药物纯度的物质统称为杂质)检测和控制,是确保药品质量、临床安全用药的前提,因此,有必要对还原型谷胱甘肽的杂质进行深入的研究。
国家食品药品监督管理局药品审评中心(CDE) 公布了药品杂质研究指导原则,ICH 对药品杂质也有相关的规定和要求药物有机杂质的鉴定限度为活性成分的0.1%,对超过该限度的杂质需进行研究,并确定该杂质在药品中的检测分析方法,有效地控制药品质量和安全性。
EUROPEAN PHARMACOPOEIA 8.0中还原型谷胱甘肽项下包含杂质A~D共四个已知杂质及一个未知杂质E,已知杂质结构如下:
杂质A 杂质B
杂质C 杂质D
在进行还原型谷胱甘肽有关物质研究的过程中,我们发现除EP杂质外,亦有一个杂质的含量随放置时间的延长而增长。在稳定性考察中发现,该杂质的增长与氧的存在有着明显的关系。由于还原型谷胱甘肽属于含巯基(-SH)的三肽化合物,易于发生水解和氧化反应,我们推测该杂质为还原型谷胱甘肽的氧化产物。通过分离纯化得到该杂质,并进行结构鉴定,结果证实该杂质为还原型谷胱甘肽的氧化产物,我们将其命名为降解产物F。
查阅相关资料发现,D. H. CALAM及S. G. WALEY于1962年首次制备了降解产物F,方法如下:将氧化型谷胱甘肽(GSSG)与硝酸银反应,滤去反应沉淀,滤液于暗处放置25小时,离心,取上清液于-10℃ 放置过夜,离心,取上清液稀释并上离子交换树脂洗脱,收集目标物洗脱液并于-10℃ 放置过夜,离心,上清液经真空干燥,得无色结晶,即The sulphinic acid(m.p. 187℃,Found:C,35.6;H,5.1;N,12.2;S,9.2;C10H17N3O8S requires C,35.4;H, 5.05;N, 12.4;S, 9.4%)。该文献从衍生物的角度出发,制备了一系列还原型谷胱甘肽的氧化产物,仅用熔点及元素组成信息对结构进行了表征,无降解产物F的核磁共振和质谱信息,不利于我们深入研究、控制产品开发过程中的杂质。
常用的杂质检测方法有自身对照法、外标法等,内控标准中还原型谷胱甘肽片及注射用还原型谷胱甘肽有关物质均采用加校正因子的高效液相色谱自身对照法进行检测。自身对照法是假设所有成分均出峰、所有杂质均与主成分分离并且所有杂质均与主成分响应因子相同的前提下,检测结果才可完全真实地表明为该物质的纯度,加校正因子克服了杂质与主成分响应因子不同的误差,使其结果更加准确,但其缺点在于需要杂质对照品进行校正因子的计算,且通过相对保留时间来对杂质进行定位亦存在一定的风险。采用已知杂质对照品法检测杂质含量,则不受杂质与主成分响应不一致等条件的影响,能对杂质进行定位并能准确测定杂质含量。
因此,制备还原型谷胱甘肽降解产物F,将其作为还原型谷胱甘肽已知杂质的对照品,能够准确有效地检测该化合物在还原型谷胱甘肽中的含量,对严格控制还原型谷胱甘肽原料及制剂的质量、提高药品的安全性具有重要的意义。
技术实现要素:
首先,本发明提供了一种还原型谷胱甘肽降解产物F的分离纯化方法。
其次,本发明提供了还原型谷胱甘肽降解产物F在还原型谷胱甘肽有关物质检测中用作杂质对照品的用途。
还原型谷胱甘肽降解产物F的分离纯化方法如下:
(1)溶解:取还原型谷胱甘肽,加水制成每1ml约含400mg的溶液。
(2)第一次分离纯化:将步骤(1)所得溶液以中压色谱分离凝胶(Middle Chromatogram Isolated GEL,MCI GEL)为固定相的自装层析柱进行初步分离,柱床直径为4~6cm,高度为60~100cm,平均粒径为70~150μm,洗脱剂为纯化水;高效液相色谱法检测洗脱流份,所用色谱柱为键合阴、阳离子配体的多模式色谱柱,流动相为pH2.5~2.9的三氟乙酸水溶液,流速为0.5~1.2ml/min,检测波长为200~210nm,根据检测结果合并洗脱流分。
其中,MCI GEL自装层析柱填料的型号选自CHP20/P70、CHP20/P120、CMG/P150,优选CHP20/P120,其对应的平均粒径为120μm;柱床直径优选5cm,高度优选80cm。所述键合阴、阳离子配体的多模式色谱柱可选Scherzo SM-C18多模式色谱柱,其直径为4.6mm,高度为150mm,粒径为3μm。
(3)第二次分离纯化:将步骤(2)所得洗脱流分以高压制备液相色谱进第二次洗脱分离,动态轴向压缩柱的填料为十八烷基硅烷键合硅胶微球,如Agilent Zorbax SB-C18,色谱柱直径21.2mm,高度250mm,粒径7μm;流动相是体积比为1:1000~10000的三氟乙酸水溶液,流速为5~12ml/min,检测波长为200~210nm,根据在线色谱图收集含有降解产物F的洗脱流分,合并洗脱流分。
(4)冷冻干燥:用冻干法处理步骤(3)所得洗脱流分,即得降解产物F。
可采用步骤(2)所述的高效液相色谱法检测所制得的还原型谷胱甘肽降解产物F的纯度。
本发明制得的还原型谷胱甘肽降解产物F,其HPLC纯度大于97%,可满足其用作杂质对照品用途的要求。在进行还原型谷胱甘肽有关物质检测时,将降解产物F作为杂质对照品,采用高效液相色谱法测定还原型谷胱甘肽中降解产物F的含量,在保证了产品在自身质量没有发生改变的前提条件下,严格了杂质的限度,提高了用药安全性,变更风险较小,有助于提高产品的质量可控性及使用安全性。
测定方法可参照以上的方法,本领域的技术人员也可根据具体情况,对色谱条件等进行调整。
附图
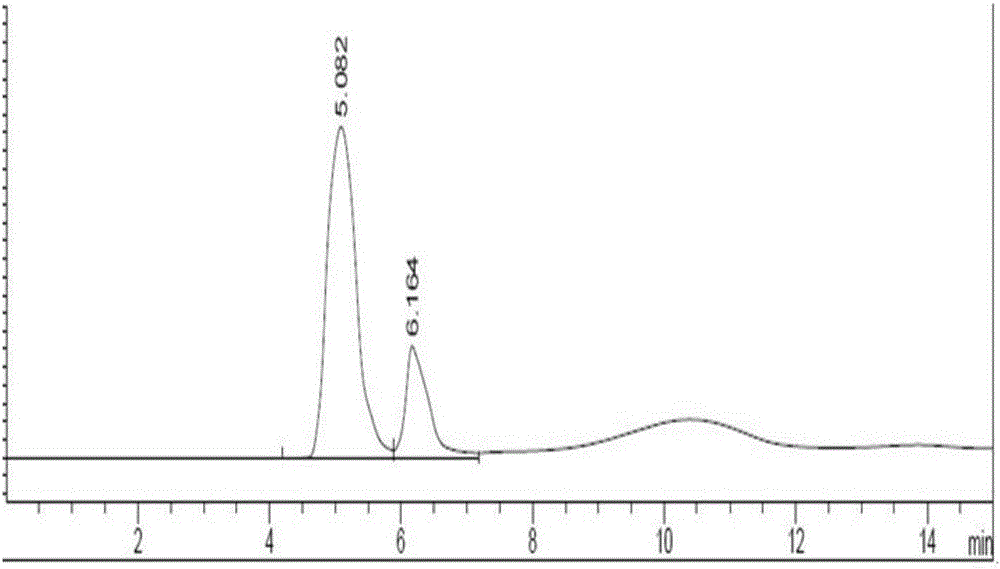
图1 第一次分离纯化所得洗脱流份中谷胱甘肽降解产物F 高效液相色谱检测图(实施例1)
图2 第二次分离纯化谷胱甘肽降解产物F高压制备液相色谱图(实施例1)
图3 第二次分离纯化所得洗脱流份中谷胱甘肽降解产物F纯度检测高效液相色谱图(实施例1)
图4 还原型谷胱甘肽样品溶液的有关物质高效液相色谱图(实施例4)
具体实施方式
下面通过实施例来进一步对本发明做出说明,但不应当被理解为是对本发明范围构成限制。
实施例 1 还原型谷胱甘肽降解产物F分离纯化方法
(1)取还原型谷胱甘肽30g,加水制成每1ml约含400mg的样品溶液;
(2)取MCI填料适量(MCI GEL型号为CHP20/P120,平均粒径120μm),用甲醇溶胀30min,将溶胀后的MCI注入玻璃层析柱中,以纯化水完全替换层析柱中甲醇(柱床直径为5cm,柱床高度为80cm);样品溶液上样后以纯化水洗脱,收集洗脱流分约20ml/份,以高效液相色谱检测洗脱流分(见图1),根据检测结果合并含降解产物F的洗脱流分;
高效液相色谱检测条件:Scherzo SM-C18柱(4.6×150mm,3μm),流动相为pH2.7的三氟乙酸水溶液,流速0.7ml/min,检测波长205nm,柱温25℃。
(3)将步骤(2)合并后的洗脱流分注入高压制备液相色谱仪,调整进样量使还原型谷胱甘肽降解产物F与其他色谱峰有较好的分离度(见图2),根据在线色谱图收集含有降解产物F的洗脱流分,合并洗脱流分,高效液相色谱法检测纯度为98.5%(见图3);
高压制备液相色谱条件:Agilent SB-C18柱(21.2×250mm,7μm),流动相为0.01%三氟乙酸水溶液,流速10ml/min;检测波长205nm。
(4)用冻干法处理步骤(3)所得洗脱流分,即得降解产物F 22.8mg。
HRMS:calcd for C10H17N3O8S, 339.0736 (M+), found 339.0759. 1H-NMR(600Hz,D2O)δppm:4.65~4.68(1H,m,7-H),3.99(2H,s,11-H),3.85~3.87(1H,m,2-H),2.82~2.86(1H,m,9-H),2.62~2.65(1H,m,9-H),2.50~2.58(2H,m,4-H),2.16~2.21(2H,m,3-H). 13C-NMR(150Hz,D2O)δppm:174.35(C-1),173.36(C-12),172.97(C-5/8),61.14(C-9),53.53(C-2),49.79(C-7),41.42(C-11),31.00(C-4),25.78(C-3)。
实施例2 还原型谷胱甘肽降解产物F分离纯化方法
(1)取还原型谷胱甘肽30g,加水制成每1ml约含400mg的样品溶液;
(2)以型号为CHP 20/P70的MCI GEL层析柱(平均粒径为70μm,柱床直径为4cm,柱床高度为60cm)对步骤(1)的样品溶液进行初步分离,洗脱剂为纯化水,收集洗脱流分约20ml/份,以高效液相色谱检测洗脱流分,根据检测结果合并含降解产物F的洗脱流分;
高效液相色谱检测色谱条件:Scherzo SM-C18柱(4.6×150mm,3μm),流动相为pH2.5的三氟乙酸水溶液,流速0.5ml/min,检测波长210nm,柱温25℃。
(3)将步骤(2)合并后的洗脱流分注入高压制备液相色谱仪,调整进样量使还原型谷胱甘肽降解产物F与其他色谱峰有较好的分离度,根据在线色谱图收集含有降解产物F的洗脱流分,合并洗脱流分,高效液相色谱法检测其纯度为97.8%;
高压制备液相色谱条件:Agilent SB-C18柱(21.2×250mm,7μm),流动相为0.1%三氟乙酸水溶液,流速5ml/min;检测波长200nm。
(4)用冻干法处理步骤(3)所得洗脱流分,即得降解产物F 18.3mg。
实施例3 还原型谷胱甘肽降解产物F分离纯化方法
(1)取注射用还原型谷胱甘肽30g,加水制成每1ml约含400mg的样品溶液;
(2)以型号为CMG20/P150的MCI GEL色谱柱(平均粒径为150μm,柱床直径为6cm,柱床高度为100cm)对步骤(1)的样品溶液进行初步分离,洗脱剂为纯化水,收集洗脱流分约20ml/份,以高效液相色谱检测洗脱流分,根据检测结果合并含降解产物F的洗脱流分;
高效液相色谱检测色谱条件:Scherzo SM-C18柱(4.6×150mm,3μm),流动相为pH2.9的三氟乙酸水溶液,流速1.2ml/min,检测波长200nm,柱温25℃。
(3)将步骤(2)合并后的洗脱流分注入高压制备液相色谱仪,调整进样量使还原型谷胱甘肽降解产物F与其他色谱峰有较好的分离度,根据在线色谱图收集含有降解产物F的洗脱流分,合并洗脱流分,高效液相色谱法检测纯度为97.1%;
高压制备液相色谱条件:Agilent SB-C18柱(21.2×250mm,7μm),流动相为0.01%三氟乙酸水溶液,流速12ml/min,检测波长210nm。
(4)用冻干法处理步骤(3)所得洗脱流分,即得降解产物F 18.7mg。
实施例4 高效液相色谱法测定还原型谷胱甘肽中降解产物F含量
色谱条件:色谱柱为十八烷基硅烷键合硅胶柱;流动相为磷酸盐缓冲溶液(取磷酸二氢钾13.6g和庚烷基磺酸钠2.2g,加水1000ml溶解后,用磷酸调节pH值至2.50±0.02)-乙腈(95:5);流速1.0ml/min;检测波长为210nm;柱温为35℃。
取装量差异下的内容物约30mg,精密称定,置25ml量瓶中(临用新配),加流动相使溶解并稀释至刻度,摇匀,作为供试品溶液;精密称取降解产物F对照品适量,用溶剂溶解并定向稀释成1ml中约含1mg的对照品溶液;取供试品溶液及对照品溶液各5μl,注入液相色谱仪,记录色谱图至主成分峰保留时间的3倍。
图4为有关物质检测图谱,其中降解产物F保留时间2.6min。从图中可以看出,降解产物F与相邻色谱峰互不干扰,在该方法中降解产物F可作为杂质对照品对还原型谷胱甘肽进行质量控制。