劣化分析方法与流程
原案申请日:2012年11月2日
原案申请号:201280053328.9(PCT/JP2012/078421)
原案申请名称:劣化分析方法
技术领域
本发明涉及一种对含有两种以上二烯系聚合物的高分子材料的各二烯系聚合物劣化状态进行分析的劣化分析方法。另外,本发明涉及一种对高分子材料的劣化状态进行分析的劣化分析方法。
背景技术:
为了对由高分子材料的劣化而引起的化学状态的变化进行评价,一般使用红外分光光度法(FT-IR)、核磁共振法(NMR)、X射线光电子分光法(XPS)等方法。虽然FT-IR或NMR能检查详细的化学状态,但由于得到的信息为全体的信息,一般难以检查从试样表面开始进展的劣化的详细化学状态。
与此相对,XPS是表面敏感的手法,因此认为对检查由劣化而引起的化学状态变化是有效的。然而,高分子材料一般大多数是混合多个聚合物制作而成,例如,在混合两种以上二烯系聚合物的材料时,检查各橡胶成分的劣化状态是重要的,但用XPS难以对各橡胶成分的劣化状态进行分析,难以对各自的劣化度进行定量。
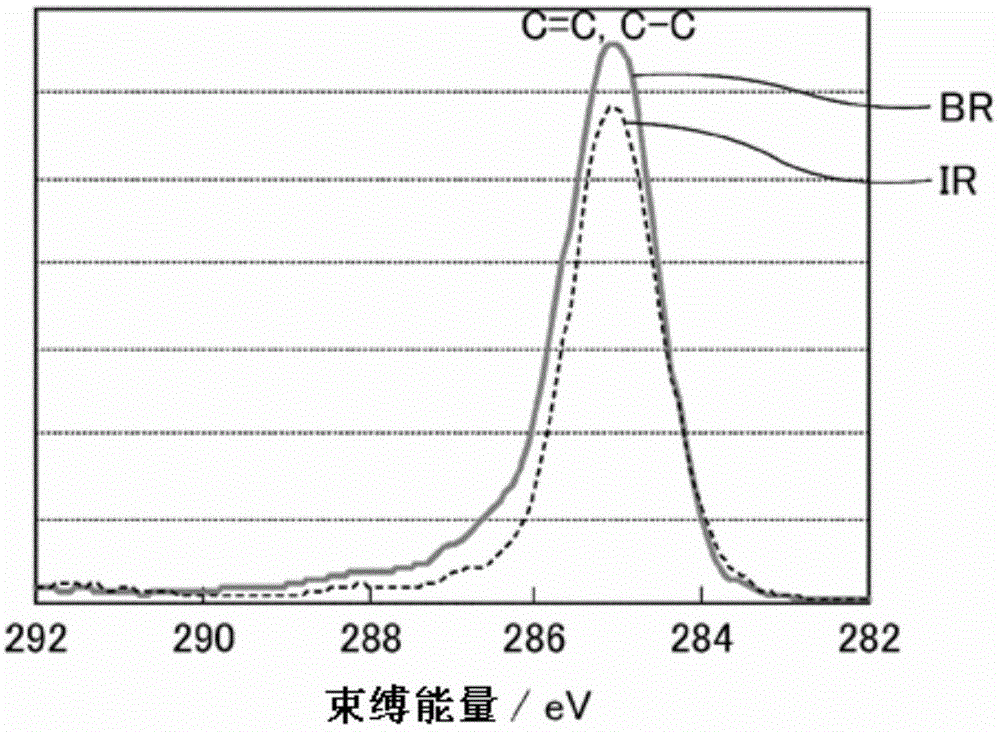
即,如图1-1所示的异戊二烯(IR)、丁二烯(BR)的各自XPS测定结果,即使在XPS测定中不同的聚合物种类也不会产生峰的化学位移,因此,难以对混合橡胶中的各成分进行详细的劣化状态分析。
另外,二烯系聚合物一般认为由劣化导致C=C(双键)切断,因此该键的检测是重要的,但C=C键(双键)的峰与C-C键(单键)的峰在285eV附近重叠,因此也不能对C=C的减少量进行定量。而且,作为二烯系聚合物的劣化原因,已知存在氧劣化和臭氧劣化,但氧劣化的峰与臭氧劣化的峰也出现重叠,因此难以对它们区别分析。
另一方面,为了对由硫交联二烯系橡胶等高分子材料的劣化而引起的化学状态变化进行评价,一般使用Swell(膨胀试验)等物性试验或红外分光光度法(FT-IR)等方法。
Swell试验是将交联的高分子材料在甲苯等低分子量溶剂中使试样膨胀而求出网链密度的方法,用该方法能检查劣化前后的橡胶分子的切断和再结合的状态,但为了查看整体变化,例如,不能对硫交联的高分子材料的高分子与硫交联中的哪个劣化正在进展进行判断。另外,FT-IR方法中,虽然能检测由劣化而生成的C=O或OH等官能团,但对S-S键的灵敏度低,因此与上述相同不能对哪个的劣化进行判断。
而且,在检查高分子材料的劣化状态方面,认为如果能知晓高分子与硫交联的各自劣化的程度的话,就确定了比以前更有效的耐劣化对策,但如上述的传统方法中,不能检查高分子与硫交联的劣化比例。
另外,关于聚合物的X射线吸收光谱,如非专利文献1~3等中所公开的,到目前为止也能被测定。然而,包括上述文献在内,没有在混合聚合物系中能检测各聚合物的劣化状态的报告。另外,也没有对该劣化状态在氧劣化与臭氧劣化中进行分离分析的报告。而且,没有通过X射线吸收光谱对劣化因素进行分离,当然也没有将X射线吸收光谱与X射线光电子分光法组合进行劣化分析的报告。
现有技术文献
非专利文献
非专利文献1:O.Dhez,H.Ade,S.G.Urquhart.J.Electron Spectrosc.Relat.Phenom.,2003,128,85-96
非专利文献2:Robert J.Klein,Daniel A.Fischer,and Joseph L.Lenhart.Langmuir.,2008,24,8187-8197
非专利文献3:冈岛敏浩,表面科学,2002年卷23,第6号,356-366
技术实现要素:
发明要解决的问题
本发明的目的在于解决上述问题并提供一种能对含有两种以上二烯系聚合物的高分子材料的劣化状态,特别是表面状态的劣化状态进行详细分析的劣化分析方法。
本发明的目的在于解决上述问题并提供一种能对含有两种以上二烯系聚合物的高分子材料的劣化状态,特别是表面状态的劣化状态,对每个二烯系聚合物进行分离并能详细分析的劣化分析方法。
本发明的目的在于解决上述问题并提供一种能对硫交联的高分子材料的劣化状态,特别是能求出高分子劣化与硫交联劣化的劣化比例的劣化分析方法。
解决问题的手段
第一的本发明涉及一种通过对含有两种以上二烯系聚合物的高分子材料照射高亮度X射线,在改变X射线的能量的同时测定X射线吸收量,来分析各二烯系聚合物的劣化状态的劣化分析方法。
上述第一的劣化分析方法中,上述高亮度X射线优选亮度为1010(光子/s/mrad2/mm2/0.1%bw)以上。另外,使用上述高亮度X射线进行扫描的能量范围优选为4000eV以下。
上述第一的劣化分析方法中,优选如下方法:通过将高亮度X射线的能量设定为260~400eV的范围对碳原子K壳层吸收端的必要范围进行扫描,根据得到X射线吸收光谱,由下述(式1-1)计算出规格化常数α和β,并在用该规格化常数α和β修正的碳原子K壳层吸收端的X射线吸收光谱中,对归属于285eV附近的π*跃迁的峰进行波形分离,用各峰面积,由下述式(1-2)求出各二烯系聚合物的劣化度。
(式1-1)
[劣化前试样测定范围的X射线吸收光谱的总面积]×α=1
[劣化后试样测定范围的X射线吸收光谱的总面积]×β=1
(式1-2)
[1-[(劣化后的各二烯系聚合物的π*的各峰面积)×β]/[(劣化前的各二烯系聚合物的π*的各峰面积)×α]]×100=劣化度(%)
上述第一的劣化分析方法,也可使用峰强度代替峰面积。
第二的本发明涉及一种通过对含有两种以上二烯系聚合物的高分子材料照射高亮度X射线,在改变X射线的能量的同时测定该高分子材料的微小区域中的X射线吸收量,来分析各二烯系聚合物的劣化状态的劣化分析方法。
上述第二的劣化分析方法中,使用上述高亮度X射线进行扫描的能量范围优选为4000eV以下。
上述第二的劣化分析方法中,优选如下方法:通过将高亮度X射线的能量设定为260~400eV的范围对碳原子K壳层吸收端的必要范围进行扫描,根据得到各二烯系聚合物的X射线吸收光谱,由下述(式2-1)计算出规格化常数α和β,并对用该规格化常数α和β修正的碳原子K壳层吸收端的X射线吸收光谱进行波形分离,用归属于所得285eV附近的π*跃迁的峰面积,由下述(式2-2)求出各二烯系聚合物的劣化度。
(式2-1)
[劣化前试样中二烯系聚合物Ai的X射线吸收光谱的总面积]×αAi=1
[劣化后试样中二烯系聚合物Ai的X射线吸收光谱的总面积]×βAi=1
(Ai表示高分子材料中所含的各二烯系聚合物)
(式2-2)
[1-[(劣化后的二烯系聚合物Ai的π*的峰面积)×βAi]/[(劣化前的二烯系聚合物Ai的π*的峰面积)×αAi]]×100=二烯系聚合物Ai的劣化度(%)
(Ai表示高分子材料中所含的各二烯系聚合物)
上述劣化分析方法(劣化度的分析方法)中,也可使用峰强度代替上述峰面积。
上述第二的劣化分析方法,优选如下方法:通过将高亮度X射线的能量设定为500~600eV的范围进行扫描,对得到的各二烯系聚合物的氧原子K壳层吸收端的X射线吸收光谱进行波形分离,将峰顶能量为532eV以上且小于532.7eV的低能量一侧的峰作为氧劣化,峰顶能量为532.7eV以上且534eV以下的范围的高能量一侧的峰作为臭氧劣化,根据下述(式2-3)计算出各二烯系聚合物的氧劣化与臭氧劣化的贡献率。
(式2-3)
[二烯系聚合物Ai的氧劣化的峰面积]/[(二烯系聚合物Ai的臭氧劣化的峰面积)+(二烯系聚合物Ai的氧劣化的峰面积)]×100=二烯系聚合物Ai的氧劣化贡献率(%)
[二烯系聚合物Ai的臭氧劣化的峰面积]/[(二烯系聚合物Ai的臭氧劣化的峰面积)+(二烯系聚合物Ai的氧劣化的峰面积)]×100=二烯系聚合物Ai的臭氧劣化贡献率(%)
(Ai表示高分子材料中所含的各二烯系聚合物)
上述劣化分析方法(氧劣化贡献率和臭氧劣化贡献率的分析方法)中,也可使用峰强度代替上述峰面积。
上述第二的劣化分析方法中,优选如下方法:根据在劣化后的各二烯系聚合物的碳原子K壳层吸收端的X射线吸收光谱,根据下述(式2-4)求出规格化常数γ,通过用该规格化常数γ并根据下述(式2-5)修正各二烯系聚合物的氧原子K壳层吸收端的X射线吸收光谱的总面积,求出氧和臭氧结合于各二烯系聚合物的量。
(式2-4)
[二烯系聚合物Ai的碳原子K壳层吸收端的X射线吸收光谱的总面积]×γAi=1
(Ai表示高分子材料中所含的各二烯系聚合物)
(式2-5)
[二烯系聚合物Ai的氧原子K壳层吸收端的X射线吸收光谱的总面积]×γAi=二烯系聚合物Ai中氧和臭氧的结合量(指数)
(Ai表示高分子材料中所含的各二烯系聚合物)
第三的本发明涉及一种通过对硫交联的高分子材料照射X射线,在改变X射线的能量的同时测定X射线吸收量而求出的高分子的劣化状态,以及通过照射一定能量的X射线,测定激发·释放的光电子而求出的硫交联的劣化状态,从而求出高分子与硫交联劣化的劣化比例的劣化分析方法。
上述第三的劣化分析方法中,作为上述高分子材料,优选为使用含有一种以上的二烯系聚合物的硫交联的高分子材料,或者将所述一种以上的二烯系聚合物与一种以上的树脂进行复合而得到的硫交联的高分子材料。
上述第三的劣化分析方法,优选如下方法:通过将X射线的能量设定为260~400eV的范围对碳原子K壳层吸收端的必要范围进行扫描,根据得到X射线吸收光谱,由下述(式3-1)计算出规格化常数α和β,对用该规格化常数α和β修正的碳原子K壳层吸收端的X射线吸收光谱进行波形分离,用归属于所得285eV附近的π*跃迁的峰面积,由下述(式3-2)求出高分子劣化度(%)。
(式3-1)
[劣化前试样测定范围的X射线吸收光谱的总面积]×α=1
[劣化后试样测定范围的X射线吸收光谱的总面积]×β=1
(式3-2)
[1-[(劣化后π*的峰面积)×β]/[(劣化前π*的峰面积)×α]]×100=高分子劣化度(%)
上述第三的劣化分析方法,优选如下方法:对通过照射一定能量的X射线而激发·释放的光电子进行分光,测定对应于硫S2p的光电子强度,得到X射线光电子能谱,对该X射线光电子能谱进行波形分离,用得到的归属于硫氧化物的峰面积由下述(式3-3)求出硫交联劣化度(%)。
(式3-3)
(归属于S2p的硫氧化物的峰面积)/(与S2p相关的总峰面积)×100=硫交联劣化度(%)
上述第三的劣化分析方法,优选如下方法:对通过照射一定能量的X射线而激发·释放的光电子进行分光,测定对应于硫S1s的光电子强度,得到X射线光电子能谱,对该X射线光电子能谱进行波形分离,用得到的归属于硫氧化物的峰面积,由下述(式3-4)求出硫交联劣化度(%)。
(式3-4)
(归属于S1s的硫氧化物的峰面积)/(与S1s相关的总峰面积)×100=硫交联劣化度(%)
此处,所使用的一定能量的X射线的能量范围优选为2.5~15keV。
再者,上述第三的劣化分析方法中,也可使用峰强度代替上述峰面积。
上述第三的劣化分析方法,优选为根据下述(式3-5)计算出高分子劣化与硫交联劣化的贡献率的方法。
(式3-5)
[高分子劣化度(%)]/[硫交联劣化度(%)]=高分子劣化与硫交联劣化的贡献率
发明效果
根据第一的本发明,因为是通过对含有两种以上二烯系聚合物的高分子材料照射高亮度X射线,在改变X射线的能量的同时测定X射线吸收量,来分析各二烯系聚合物的劣化状态的劣化分析方法,所以,即使对于含有两种以上二烯系聚合物的高分子材料,也能对每个二烯系聚合物的劣化状态,特别是表面状态的劣化状态进行详细分析。因此,关于含有两种以上二烯系聚合物的高分子材料的劣化,可以测定每个二烯系聚合物的劣化度(%)。
根据第二的本发明,因为是通过对含有两种以上二烯系聚合物的高分子材料照射高亮度X射线,在改变X射线的能量的同时测定该高分子材料的微小区域中的X射线吸收量,来分析各二烯系聚合物的劣化状态的劣化分析方法,所以,即使对于含有两种以上二烯系聚合物的高分子材料,也能对每个二烯系聚合物的劣化状态,特别是表面状态的劣化状态进行详细分析。因此,关于含有两种以上二烯系聚合物的高分子材料的劣化,可以对每个二烯系聚合物进行劣化度(%)、氧劣化与臭氧劣化的贡献率,以及氧和臭氧的结合量进行测定。
根据第三的本发明,因为是通过对硫交联的高分子材料照射X射线,在改变X射线能量的同时测定X射线吸收量而求出的高分子的劣化状态,以及通过照射一定能量的X射线,测定激发·释放的光电子而求出的硫交联的劣化状态,从而求出高分子与硫交联劣化的劣化比例的劣化分析方法,所以,对硫交联的高分子材料的劣化状态,特别是能求出高分子劣化与硫交联劣化的劣化比例。因而,能辨别高分子劣化、硫交联劣化中的哪个劣化更进展,并能谋求与比以前更有效的耐劣化对策。
附图说明
图1-1示出了异戊二烯橡胶和丁二烯橡胶的碳的1s轨道中的XPS测定结果图。
图1-2示出了了异戊二烯橡胶和丁二烯橡胶的碳原子K壳层吸收端附近的NEXAFS测定结果图。
图1-3示出了混合了异戊二烯橡胶和丁二烯橡胶的橡胶新品、施行臭氧劣化1小时的劣化品的碳原子K壳层吸收端的NEXAFS测定结果图(规格化前)。
图1-4示出了混合了异戊二烯橡胶和丁二烯橡胶的橡胶新品、施行臭氧劣化1小时的劣化品的碳原子K壳层吸收端的NEXAFS测定结果的图(规格化后)。
图1-5是对于混合了异戊二烯橡胶和丁二烯橡胶的橡胶新品,对规格化后的碳原子K壳层吸收端的NEXAFS测定结果进行波形分离的图。
图1-6是对于混合了异戊二烯橡胶和丁二烯橡胶的橡胶施行臭氧劣化1小时的劣化品,对规格化后的碳原子K壳层吸收端的NEXAFS测定结果进行波形分离的图。
图2-1是高分子材料中的微XAFS法与通常的XAFS法的测定范围的示意图。
图2-2是在进行了暴露试验的高分子材料的氧原子K壳层的σ峰中的图像。
图2-3示出了对于IR与SBR的混合橡胶的新品、施行臭氧劣化7小时的劣化品、施行氧劣化1周的劣化品的试样,通过XPEEM法的IR的碳原子K壳层吸收端的测定结果图(规格化前)。
图2-4示出了对于IR与SBR的混合橡胶的新品、施行臭氧劣化7小时的劣化品、施行氧劣化1周的劣化品的试样,通过XPEEM法的IR的碳原子K壳层吸收端的测定结果图(规格化后)。
图2-5示出了对于IR与SBR的混合橡胶的新品、施行臭氧劣化7小时的劣化品、施行氧劣化1周的劣化品的试样,通过XPEEM法的IR的氧原子K壳层吸收端的测定结果图。
图2-6示出了对于IR与SBR的混合橡胶进行了复合劣化(氧劣化与臭氧劣化)的劣化品的试样,通过XPEEM法的IR的氧原子K壳层吸收端的测定结果图。
图2-7示出了对于IR与SBR的混合橡胶施行臭氧劣化7小时的劣化品、施行臭氧劣化1小时的劣化品的试样,通过XPEEM法的IR的测定结果图(规格化后)。
图3-1示出了天然橡胶与丁二烯橡胶的混合橡胶的新品和施行臭氧劣化7小时的试样的碳原子K壳层吸收端的NEXAFS测定结果图(规格化前)。
图3-2示出了丁二烯橡胶新品和施行臭氧劣化7小时的试样的碳原子K壳层吸收端的NEXAFS测定结果图(规格化后)。
图3-3示出了天然橡胶与丁二烯橡胶的混合橡胶的新品和进行热氧劣化1周的试样的高分子材料的S2p(硫的2p轨道)中的X射线光电子能谱的测定结果图。
图3-4示出了天然橡胶与丁二烯橡胶的混合橡胶的新品和进行热氧劣化1周的试样的高分子材料的S1s(硫的1s轨道)中的X射线光电子能谱的测定结果图。
具体实施方式
第一的本发明的劣化分析方法是一种通过对含有两种以上二烯系聚合物的高分子材料照射高亮度X射线,在改变X射线能量的同时测定X射线吸收量,来分析各二烯系聚合物的劣化状态的方法。作为橡胶等高分子材料的劣化因素,已知由紫外线、氧、臭氧、热等引起聚合物分子链的劣化、交联结构的劣化等,但为了改善耐劣化性,重要的是知晓由什么因素引起聚合物分子链、交联结构的怎样变化。
关于此点,上述的第一劣化分析方法着眼于使用高亮度X射线,其方法是:对于新品和劣化后的含有两种以上二烯系聚合物的高分子材料,各自在改变高亮度X射线的能量的同时进行照射,测定X射线吸收量,得到各二烯系聚合物的各光谱,并对这些光谱进行比较,由此能对劣化后的高分子材料中的各二烯系聚合物的劣化状态进行分析。
具体而言,可采用以高亮度X射线测定关注的特定元素的吸收端附近的X射线吸收光谱(NEXAFS(吸收端附近X射线吸收微细结构):Near Edge X-ray Absorption Fine Structure)的手法,由于软X射线具有轻元素吸收,因此能详细分析软材料的化学状态。
由于NEXAFS法使用X射线能量进行扫描,因此光源中需要连续X射线发生装置,为了分析详细的化学状态中,需要测定高S/N比和S/B比的X射线吸收光谱。因此,从同步加速器放射的X射线至少具有1010(光子/s/mrad2/mm2/0.1%bw)以上的亮度并且是连续的X射线源,因而最适于NEXAFS测定。另外,bw表示从同步加速器放射的X射线的带宽。
上述第一的劣化分析方法中,上述高亮度X射线的亮度(光子/s/mrad2/mm2/0.1%bw)优选为1010以上,更优选为1011以上,进一步优选为1012以上。虽然对上限并没有特别限定,但优选为使用无放射线损伤程度以下的X射线强度。
另外,上述第一劣化分析方法中,上述高亮度X射线的光子数(光子/秒)优选为107以上,更优选为109以上。虽然对上限并没有特别限定,但优选为使用无放射线损伤程度以下的X射线强度。
上述第一的劣化分析方法中,使用上述高亮度X射线进行扫描的能量范围优选为4000eV以下,更优选为1500eV以下,进一步优选为1000eV以下。如果超过4000eV,则可能无法分析作为目标高分子复合材料中的劣化状态。对下限并没有特别限定。
例如,测定可通过以下方法实施:通过对设置于超高真空中的试样照射软X射线使光电子逸出,为了补偿该光电子而从接地端电子流入,再测定该试样电流。因此,虽然表面敏感,但由于作为可测定的条件可举出在真空中不产生气体和具有导电性,因而至今为止以结晶或分子吸附的研究为主,几乎不进行对产生气体且为绝缘体的橡胶试样的研究。
然而,由于作为同样表面敏感手法的ESCA查看的是原子的内壳层,因此难以详细分离出高分子的劣化状态,与此相对,由于NEXAFS查看的原子与原子相互影响的外壳层,因此与ESCA相比,受到所考察元素上结合的元素的影响大,因此认为能分离出各个分子状态,劣化因素的分离成为可能,从而完成第一的本发明。
具体而言,可用下述方法实施。
将试样安装在试样架后,设置于用于实施X射线吸收测定的真空室中。然后,用分光器对从同步加速器放射的连续X射线进行单色化,照射试样。此时,二次电子·光电子从试样表面逸出至真空中,但为了补偿失去的电子,电子从接地端得到补充。此处,将从接地端流入的电流作为X射线吸收强度I,将设置在束线的光学系统中的金属网电流作为入射X射线强度I0,由下述(公式)求出X射线吸收量μL(电子产额法)。另外,本手法虽然可适用于Lanbert-Beer公式,但在电子产额法的情况下,可认为下述(公式)近似成立。
[数1]
(公式)
I0(E)/I(E)=exp(μL)≌μL(E:X射线的能量,L:试样厚度,μ:吸收系数)
NEXAFS的测定方法中,可代表性地使用以下三种方法。本发明的实施例中,虽然用电子产额法实施,但并不限定于此,可使用各种检测方法,也可以进行组合同时测量。
(透过法)
是检测能透过试样的X射线强度的方法。透过光强度测定中,可使用光电二极管阵列检测器等。
(荧光法)
是对试样照射X射线时产生的荧光X射线进行检测的方法。在上述透过法的情况下,如果对试样中的含量少的元素进行X射线吸收测定,则不仅信号弱且因含量多的元素的X射线吸收引起背景变强,因此成为S/B比不好的光谱。与此相对,荧光法(特别是使用能量分散型检测器等的情况下)中,由于可仅测定来自目标元素的荧光X射线,因此受含量多的元素影响小。因此,在对含量少的元素进行X射线吸收光谱测定的情况下是有效的。另外,由于荧光X射线透过力强(与物质的相互作用小),因此可以检测试样内部产生的荧光X射线。所以,本手法是继透过法之后作为获得全体信息的方法是最适合的。
(电子产额法)
是对试样照射X射线时流出的电流进行检测的方法。因此需要试样为导电物质。由于高分子材料是绝缘体,因此至今为止的高分子材料的X射线吸收测定中绝大部分使用通过蒸镀或旋涂等将试样承载在非常薄的基板上的形成物,但本发明中,通过用薄切片机将高分子材料加工(切割)为100μm以下,优选为10μm以下,更优选为1μm以下,进一步优选为500nm以下,可实现S/B比和S/N比高的测定。
另外,作为电子产额法的特征,可举出表面敏感(试样表面数nm水平的信息)的方面。虽然如果对试样照射X射线则电子从元素中逸出,但由于电子与物质间的相互作用强,因此在物质中的平均自由行程短。
通过使用上述电子产额法对高分子材料的X射线吸收光谱测定进行分析,能分析劣化度(%)。以下,对此进行说明。
作为上述第一的劣化分析方法,例如,可举出如下方法:通过将上述高亮度X射线的能量为260~400eV的范围对碳原子K壳层吸收端的必要范围进行扫描,根据得到X射线吸收光谱,由下述(式1-1)计算出规格化常数α和β,对用该规格化常数α和β修正的碳原子K壳层吸收端的X射线吸收光谱中归属于285eV附近的π*跃迁的峰进行波形分离,用各峰面积由下述式(1-2)求出各二烯系聚合物的劣化度。
(式1-1)
[劣化前试样测定范围的X射线吸收光谱的总面积]×α=1
[劣化后试样测定范围的X射线吸收光谱的总面积]×β=1
(式1-2)
[1-[(劣化后各二烯系聚合物的π*的各峰面积)×β]/[(劣化前各二烯系聚合物的π*的各峰面积)×α]]×100=劣化度(%)。
由此,能得到劣化后的各二烯系聚合物的劣化度(%),并能分析劣化率。此处,上述求出劣化度的方法中,将上述高亮度X射线的能量优选为260~350eV的范围。再者,上述求出劣化度的方法中,在进行上述(式1-1)的操作前,根据吸收端前的斜率进行评价并减去背景。
上述求出劣化度的方法中,在上述(式1-1)中X射线吸收光谱的总面积是对测定范围内的光谱进行积分得到的,并能根据测定条件等改变能量范围。
关于上述求出劣化度的方法,使用IR与BR混合的橡胶新品、施行臭氧劣化1小时的劣化品的试样的例子,进行具体说明。
预先进行IR和BR的各自碳原子K壳层吸收端附近的NEXAFS测定。其测定结果如图1-2所示,IR和BR都有归属于285eV附近的C=C(双键)的π*跃迁峰(也被称为π*(C=C)峰),但IR的峰顶能量为285.4eV附近,BR的峰顶能量为284.9eV附近,并根据二烯系聚合物的种类(不同分子),显示不同的峰顶能量。
再者,图1-2中示出了IR和BR的例子,但第一的本发明适用于混合聚合物的各自的峰顶能量不同的情况,特别是不限定聚合物种类,例如,苯乙烯-丁二烯橡胶(SBR)的峰顶能量为285.0eV附近,同样地,由于能量不同,即使在含有SBR的混合橡胶也采用同样的手法。另外,由于天然橡胶(NR)具有与IR相同的峰顶能量,因此可与IR相同的方式进行处理。
新品、劣化品(臭氧劣化)的试样的碳原子K壳层吸收端的NEXAFS测定结果如图1-3所示。如图1-3所示的,虽然劣化的试样在285eV附近的π*峰与新品相比变小,但用NEXAFS法难以进行绝对值测定。其理由是,从光源到试样的距离等微妙变化会对X射线吸收光谱的大小产生影响。根据以上理由,对于根据碳原子K壳层吸收端的XPEEM法或STXM法的测定结果,不能进行试样间的简单比较。
因此,为了比较测定的试样间的X射线吸收光谱,进行如下方式的规格化(为了能直接比较而修正各试样的X射线吸收光谱)。由于劣化前后的碳壳层的X射线吸收量不变,因此用上述(式1-1),使碳原子K壳层吸收端的峰面积规格化为1。即,首先对于规格化前的X射线吸收光谱,根据上述(式1-1)计算出规格化常数α、β,接着通过对规格化前的X射线吸收光谱乘以α、β的光谱进行修正(规格化),能直接比较试样间的π*峰。由此得到的规格化后的碳原子K壳层吸收端的光谱如图1-4所示。
对于新品、劣化品的各自试样,将图1-4的规格化后的碳原子K壳层吸收端光谱的π*(C=C)峰波形分离为归属于峰顶能量285.4eV附近的IR和峰顶能量284.9eV附近的BR的各峰。图1-5~1-6示出了对于新品、劣化品的各自波形分离为IR和BR的图。再者,作为用于分离成归属于各聚合物成分的各峰的波形分离方法,可使用高斯(Gauss)函数。另外,根据需要也可使用罗伦兹(Lorenz)函数、或者高斯(Gauss)函数与罗伦兹(Lorenz)函数权重总和的任意函数。
使用归属于图1-5的新品、图1-6的劣化品的IR的各自峰面积,通过用(式1-2)来确定IR的劣化度。对于BR也同样地,通过用(式1-2)来确定BR的劣化度。此处,劣化度是指从劣化前至劣化后的归属于各二烯系聚合物的π*的各峰减少率,表示试样中所混合的各二烯系聚合物的劣化率(%)。
再者,上述求出劣化度的方法中,即使在上述(式1-2)中使用峰强度代替峰面积,同样也能求出劣化度。
另外,虽然上述对于臭氧劣化品进行了说明,但即使对于氧劣化品、以臭氧与氧这两者方式进行劣化的劣化品,也能以同样的手法进行分析,能求出每个聚合物成分的劣化度。
上述第一的本发明方法,例如,可通过佐贺县立九州同步加速器光研究中心的BL12束线进行。
另外,作为适用于第一的本发明的上述高分子材料,只要是含有两种以上二烯系聚合物的材料的话,就没有特别限定,例如,适宜使用含有两种以上二烯系聚合物的混合橡胶材料,该混合橡胶材料与一种以上的树脂进行复合的复合材料。作为上述二烯系聚合物,可举出天然橡胶(NR)、异戊二烯橡胶(IR)、丁二烯橡胶(BR)、苯乙烯-丁二烯橡胶(SBR)、丙烯腈-丁二烯橡胶(NBR)、氯丁橡胶(CR)、丁基橡胶(IIR)、卤化丁基橡胶(X-IIR)、苯乙烯-异戊二烯-丁二烯橡胶(SIBR)等具有双键的聚合物。
作为上述树脂,没有特别限定,例如,可举出橡胶工业领域中通用的树脂,例如,可举出C5系脂肪族石油树脂、环戊二烯系石油树脂等石油树脂。本发明的劣化分析方法可适用于这些材料。
如上所述,对于混合了多种二烯系聚合物的新品和劣化后的高分子材料,通过在改变高亮度X射线的能量的同时进行照射,从其X射线吸收量分析劣化度,不仅对整体试样,而且还能对各二烯系聚合物的劣化状态进行分析。因此,通过使用第一的本发明的手法,对于一般使用的混合材料,可进行每个聚合物成分的劣化度分析。
接着,第二的本发明的劣化分析方法是一种通过对含有两种以上二烯系聚合物的高分子材料照射高亮度X射线,在改变X射线的能量的同时测定该高分子材料的微小区域中的X射线吸收量,来分析各二烯系聚合物的劣化状态的方法。
如上所述,作为橡胶等高分子材料的劣化因素,已知由紫外线、氧、臭氧、热等引起聚合物分子链的劣化、交联结构的劣化等,但为了改善耐劣化性,重要的是知晓由什么因素引起聚合物分子链、交联结构的怎样变化。
本发明人提出了利用日本专利特愿2011-167131中的使用高亮度X射线对特定元素的吸收端附近的X射线吸收光谱进行测定的NEXAFS(Near Edge X-ray Absorption Fine Structure)法,求出高分子材料的劣化度、氧劣化贡献率、臭氧劣化贡献率,以及氧和臭氧的结合量。然而,虽然该方法能获得对于整体高分子材料的信息,但在混合了多个二烯系聚合物的高分子材料的情况下,无法对各二烯系聚合物的劣化状态进行分离分析。另外,上述第一的本发明的劣化分析方法中,虽然提出了利用NEXAFS法来评价高分子材料中所混合的各二烯系聚合物的劣化度的方法,但该方法也无法测定每个二烯系聚合物中的氧劣化与臭氧劣化的贡献率,以及氧和臭氧的结合量。
与此相对,第二的本发明的劣化分析方法中,通过对新品和劣化后的含有两种以上二烯系聚合物的高分子材料分别在改变高亮度X射线的能量的同时进行照射,测定该高分子材料的微小区域中的X射线吸收量,得到各二烯系聚合物的各光谱,并对这些光谱进行比较,能对劣化后的高分子材料中的各二烯系聚合物的劣化状态进行分离分析。即,在第二的本发明的劣化分析方法对每个二烯系聚合物进行X射线吸收光谱进行测定,并对得到的光谱进行比较。因此,不仅对劣化度(%),而且还能对每个二烯系聚合物的氧劣化与臭氧劣化的贡献率,以及氧和臭氧的结合量进行测定。
具体而言,可采用作为使用高亮度X射线测定试样的微小区域中的X射线吸收光谱方法的微XAFS(X-ray Absorption Fine Structure)。如图2-1所示的,通常的XAFS由于不具有空间分辨率,因此能检测整体试样的吸收量,与此相对,微XAFS是一种对试样的微小区域中的X射线吸收光谱进行测定的测定手法,通常具有100nm以下左右的空间分辨率。因此,通过采用微XAFS,能对试样中所混合的每个二烯系聚合物的X射线吸收光谱进行测定,检测各二烯系聚合物的不同吸收量。
从空间分辨率优异的方面考虑,优选为作为软X射线区域中的测定方法(微NEXAFS)的微XAFS,更优选为X射线光电子显微镜(XPEEM:X-ray Photo emission electron microscopy)法或扫描型透射X射线显微镜(STXM:Scanning Transmission X-ray Microscopy)法。
XPEEM法虽然通过对试样照射高亮度X射线,试样吸收X射线而从试样表面释放出电子,但由于释放出的电子保持其位置和强度直接通过透镜(静电型或磁场型)进行扩大,因此如图2-2所示的,能对混合了两种以上二烯系聚合物状态进行形态观察。另外,来自试样的电子释放量与X射线的吸收系数成比例。因此,能检测各二烯系聚合物的不同吸收量,并用上述(式2-1)~(式2-5),能求出各二烯系聚合物的劣化度、氧劣化与臭氧劣化的贡献率,以及氧和臭氧的结合量。
再者,XPEEM法还可通过对试样照射高亮度X射线,在X射线周围对测定试样进行光栅扫描,将光电子释放强度作为试样表面位置的函数进行测定。
STXM法通过对试样的微小区域照射用菲涅尔区板聚光的高亮度X射线并测定穿过试样的光(透过光)与入射光,能测定微小区域的X射线吸收量。因此,能检测各二烯系聚合物的不同吸收量,并用上述(式2-1)~(式2-5),能求出各二烯系聚合物的劣化度、氧劣化与臭氧劣化的贡献率,以及氧和臭氧的结合量。
再者,还可代替菲涅尔区板,通过用X射线反射镜的Kirkpatrick-Baez(K-B)聚光系统对高亮度X射线进行聚光。
XPEEM法或STXM法用于以X射线能量进行扫描的光源需要连续X射线产生装置,并且详细的化学状态分析中需要测定高S/N比和S/B比的X射线吸收光谱。因此,从同步加速器放射的X射线由于具有至少1010(光子/s/mrad2/mm2/0.1%bw)以上的亮度并且是连续的X射线源,因而,最适于通过XPEEM法或STXM法的测定。另外,bw表示从同步加速器放射的X射线的带宽。
上述第二的劣化分析方法中,优选上述高亮度X射线的亮度、高亮度X射线的光子数、使用高亮度X射线进行扫描的能量范围与上述第一的劣化分析方法相同。
作为表面敏感手法的ESCA,由于可查看原子的内壳层而难以详细地对高分子的劣化状态进行分离,与此相对,XPEEM法或STXM法,由于可查看向非占有轨道的激发,与ESCA相比,很大程度上受到结合于考查元素的元素影响,因此,认为能分离各个结合状态进行并能进行劣化因素的分离,从而完成第二的本发明。
通过使用上述微XAFS法对高分子材料的X射线吸收光谱测定进行分析,能对所混合的每个聚合物进行劣化度(%)、氧劣化与臭氧劣化的贡献率(%)、氧和臭氧的结合量(劣化指标)的分析。以下,对此进行说明。
作为第二的劣化分析方法,例如,可举出如下方法:通过将上述高亮度X射线的能量为260~400eV的范围对碳原子K壳层吸收端的必要范围进行扫描,根据得到各二烯系聚合物的X射线吸收光谱,由下述(式2-1)计算出规格化常数α和β,对用该规格化常数α和β修正的碳原子K壳层吸收端的X射线吸收光谱进行波形分离,用得到的归属于285eV附近的π*跃迁的峰面积,由下述(式2-2)求出各二烯系聚合物的劣化度。
(式2-1)
[劣化前试样中二烯系聚合物Ai的X射线吸收光谱的总面积]×αAi=1
[劣化后试样中二烯系聚合物Ai的X射线吸收光谱的总面积]×βAi=1
(Ai表示高分子材料中所含的各二烯系聚合物)
(式2-2)
[1-[(劣化后二烯系聚合物Ai的π*的峰面积)×βAi]/[(劣化前二烯系聚合物Ai的π*的峰面积)×αAi]]×100=二烯系聚合物Ai的劣化度(%)
(Ai表示高分子材料中所含的各二烯系聚合物)
由此,可得到劣化后的各二烯系聚合物的劣化度(%),并能分析劣化率。此处,上述求出劣化度的方法中,优选为将上述高亮度X射线的能量设为260~350eV的范围。再者,上述求出劣化度的方法在进行上述(式2-1)的操作前,根据吸收端前的斜率进行评价并减去背景。
上述求出劣化度的方法中,上述(式2-1)中X射线吸收光谱的总面积是对测定范围内的光谱进行积分得到的,并能根据测定条件等改变能量范围。
关于上述求出劣化度的方法,以IR与SBR的混合橡胶的新品、施行臭氧劣化7小时的劣化品、施行氧劣化1周的劣化品的试样的测定结果作为例子,进行具体说明。
对于IR与SBR的混合橡胶的新品、施行臭氧劣化7小时的劣化品、施行氧劣化1周的劣化品的试样,通过XPEEM法的IR的碳原子K壳层吸收端的测定结果如图2-3所示。如图2-3所示的劣化试样中的285eV附近的π*峰与新品相比变小,难以使用XPEEM法或STXM法进行绝对值测定。其理由是,从光源到试样的距离等微妙变化会对X射线吸收光谱的大小产生影响。根据以上理由,对于根据碳原子K壳层吸收端的XPEEM法或STXM法的测定结果,不能进行试样间的简单比较。
因此,为了比较测定的试样间的X射线吸收光谱,进行如下方式的规格化(为了能直接比较而修正各试样的X射线吸收光谱)。由于劣化前后中碳原子的K壳层的X射线吸收量不变,用上述(式2-1),将碳原子K壳层吸收端的峰面积规格化为1。即,首先对于规格化前的X射线吸收光谱,根据上述(式2-1)计算出规格化常数α、β,接着,通过对规格化前的X射线吸收光谱中乘以α、β的光谱进行修正(规格化),能直接比较试样间的π*峰。
由此得到的规格化后的碳原子K壳层吸收端的光谱如图2-4所示。根据规格化的光谱,用上述(式2-2)确定劣化度。此处,劣化度是归属于从劣化前至劣化后的各二烯系聚合物的π*的各峰减少率,并表示试样中所混合的各二烯系聚合物的劣化率(%)。
通过对SBR也进行上述求出劣化度的方法,能分离分析混合橡胶中的IR与SBR的劣化度。
再者,上述求出劣化度的方法中,即使在上述(式2-2)中使用峰强度代替峰面积,同样也能求出劣化度。另外,即使使用STXM法代替XPEEM法,同样也能求出劣化度。
另外,作为上述第二的劣化分析方法,可举出如下方法:通过将上述高亮度X射线的能量为500~600eV的范围进行扫描,对得到各二烯系聚合物的氧原子K壳层吸收端的X射线吸收光谱进行波形分离,将峰顶能量为532eV以上且小于532.7eV的低能量一侧的峰作为氧劣化,峰顶能量为532.7eV以上且534eV以下范围的高能量一侧的峰作为臭氧劣化,由下述(式2-3)计算出各二烯系聚合物的氧劣化与臭氧劣化的贡献率。
(式2-3)
[二烯系聚合物Ai氧劣化的峰面积]/[(二烯系聚合物Ai臭氧劣化的峰面积)+(二烯系聚合物Ai氧劣化的峰面积)]×100=二烯系聚合物Ai的氧劣化贡献率(%)
[二烯系聚合物Ai臭氧劣化的峰面积]/[(二烯系聚合物Ai臭氧劣化的峰面积)+(二烯系聚合物Ai氧劣化的峰面积)]×100=二烯系聚合物Ai的臭氧劣化贡献率(%)
(Ai表示高分子材料中所含的各二烯系聚合物)
由此,可得到劣化后的高分子材料中的各二烯系聚合物的氧劣化与臭氧劣化的贡献率(%),并能分析各自的劣化因素。
再者,上述计算出贡献率方法中,在进行上述(式2-3)的操作前,根据吸收端前的斜率进行评价并减去背景。
关于上述计算出贡献率方法,以IR与SBR的混合橡胶的新品、施行臭氧劣化7小时的劣化品、施行氧劣化1周的劣化品的试样的测定结果作为例子,进行具体说明。
首先,对于IR与SBR的混合橡胶的新品、施行臭氧劣化7小时的劣化品、施行氧劣化1周的劣化品的试样,根据XPEEM法的IR的氧原子K壳层吸收端的测定结果如图2-5所示。如图所示的,明确了臭氧劣化的试样具有532.7eV以上且534eV以下的峰,氧劣化的试样具有532eV以上且小于532.7eV的峰,两个峰中的高能量一侧的峰归属于臭氧劣化,低能量一侧的峰归属于氧劣化。
而且,图2-6示出了对于进行复合劣化(氧劣化与臭氧劣化)的劣化品的试样,根据XPEEM法的IR的氧原子K壳层吸收端的测定结果。如图2-6所示的,在532~534eV中,检测到具有双肩状的峰。其认为是由氧劣化引起的低能量一侧的峰(532eV以上且小于532.7eV)与由臭氧劣化引起的高能量一侧的峰(532.7eV以上且534eV以下)重叠而造成的。因此,进行峰分离后,用上述(式2-3)确定氧劣化与臭氧劣化的贡献率。由此,对于氧劣化与臭氧劣化同时进展的试样,能对两劣化因素中的氧劣化比例和臭氧劣化比例进行分析。
通过对SBR也进行上述计算出贡献率的方法,能对混合橡胶中的各自IR与SBR进行氧劣化与臭氧劣化的贡献率分析。
再者,上述计算出贡献率的方法中,即使在上述(式2-3)中使用峰强度代替峰面积,同样也能求出氧劣化与臭氧劣化的贡献率。另外,即使使用STXM法代替XPEEM法,同样也能求出氧劣化与臭氧劣化的贡献率。
而且,作为上述第二的劣化分析方法,也可举出如下方法:根据劣化后的各二烯系聚合物的碳原子K壳层吸收端的X射线吸收光谱,由下述(式2-4)求出规格化常数γ,用该规格化常数γ由下述(式2-5)对各二烯系聚合物的氧原子K壳层吸收端的X射线吸收光谱的总面积进行修正,来求出氧和臭氧结合于各二烯系聚合物的量。
(式2-4)
[二烯系聚合物Ai的碳原子K壳层吸收端的X射线吸收光谱的总面积]×γAi=1
(Ai表示高分子材料中所含的各二烯系聚合物)
(式2-5)
[二烯系聚合物Ai的氧原子K壳层吸收端的X射线吸收光谱的总面积]×γAi=二烯系聚合物Ai中氧和臭氧的结合量(指数)
(Ai表示高分子材料中所含的各二烯系聚合物)
由此,能测定由劣化引起的结合于各二烯系聚合物的氧和臭氧量,并将其作为劣化指标。
上述求出结合量的方法中,光谱的总面积是对测定范围内的光谱进行积分得到的,并能根据测定条件等改变能量范围。
关于上述求出结合量的方法,以IR与SBR的混合橡胶施行臭氧劣化7小时的劣化品、施行臭氧劣化1小时的劣化品的试样的测定结果作为例子,进行具体说明。
关于上述试样,根据XPEEM法的IR的测定结果如图2-7所示。其结果是:根据碳原子K壳层吸收端的X射线吸收光谱,用上述(式2-4)求出规格化常数γ,并用(式2-5)与上述同样地进行规格化。规格化的氧原子K壳层吸收端的峰面积认为是氧和臭氧的结合量。如图2-7所示的,由于进行7小时劣化后的试样面积大于进行1小时劣化后的试样面积,因此,该值可作为劣化指数。该劣化指数的数字越大,表示结合于IR的氧和臭氧的量越多。因此,根据氧原子K壳层吸收端的峰面积增加率,可测定由氧和臭氧结合于IR引起的劣化率。
通过对SBR也进行上述计算出劣化率的方法,能对混合橡胶中的各自IR与SBR进行氧和臭氧的结合量的分析。
再者,即使使用STXM法代替XPEEM法,同样也能对氧和臭氧的结合量进行分析。
上述第二的本发明的方法,例如,如果是XPEEM法,则可使用Spring-8BL17SU束线附带的分光型光电子/低能量电子显微镜(SPELEEM:Elmitec公司制造)进行实施,如果是STXM法,则可使用美国Lawrence Berkeley国立研究所的Advanced Light Source(ALS)的束线5.3.2进行实施。
另外,作为适用于第二的本发明的上述高分子材料,只要是含有两种以上二烯系聚合物的材料的话,就没有特别限定,可使用与上述第一的本发明相同的材料等。第二的本发明的劣化分析方法可适用于这些材料。
如上所述,对于混合了多种二烯系聚合物的新品和劣化后的高分子材料,通过在改变高亮度X射线的能量的同时进行照射,并根据该高分子材料的微小区域中的X射线吸收量进行劣化度的分析,从而对各二烯系聚合物的劣化状态进行分析。因此,通过使用第二的本发明的手法,对于一般使用的混合材料,能进行每个聚合物成分的劣化度分析,有助于难以劣化的材料开发。
接着,第三的本发明的劣化分析方法是一种如下方法:通过对硫交联的高分子材料照射X射线,在改变X射线的能量的同时测定X射线吸收量而求出高分子的劣化状态,以及通过照射一定能量的X射线、测定激发·释放的光电子而求出硫交联的劣化状态,从而求出高分子裂化与硫交联劣化的劣化比例。
作为硫化橡胶等硫交联的高分子材料的劣化因素,已知由紫外线、氧、臭氧、热等引起的聚合物分子链的劣化、硫交联的劣化等,但为了改善耐劣化性,重要的是知晓由什么因素引起聚合物分子链、硫交联结构怎样的变化。
关于这方面,第三的本发明的劣化分析方法首先着眼于使用X射线,通过对新品和劣化后的高分子材料分别在改变X射线的能量的同时照射X射线,测定X射线吸收量而得到各光谱,并对各光谱进行比较,分析劣化后的高分子材料的劣化状态。具体而言,使用测定特定元素的吸收端附近的X射线吸收光谱(NEXAFS:(吸收端附近X射线吸收微结构)Near Edge X-ray Absorption Fine Structure)的手法等进行试样的测定,根据碳原子K壳层吸收端的X射线吸收光谱的峰面积等,能对聚合物部分的劣化状态进行分析。
如果使用NEXAFS法,则虽然根据氧K壳层吸收端附近的X射线吸收光谱也能对高分子材料中的氧或臭氧等的结合量进行分析,但用该方法无法判断是结合于高分子部分还是结合于硫交联部分。因此,第三的本发明中,在通过使用X射线对高分子(聚合物分子链)部分的劣化状态进行分析的同时,也能通过测定照射一定能量的X射线而激发·释放的光电子对硫交联的劣化状态进行分析。具体而言,使用X射线光电子分光法(XPS:X-ray Photoelectron Spectroscopy)等进行试样的测定,可根据归属于硫的峰面积等对硫交联的劣化状态进行分析。因此,通过采用第三的本发明的方法,可测定高分子和硫交联各自的劣化度,能分析哪个劣化更进展,即能分析劣化比例。
第三的本发明中,使用NEXAFS法照射X射线,在改变X射线的能量的同时测定X射线吸收量时,由于软X射线具有轻元素吸收,因此能详细分析软材料的化学状态。
可通过与上述第一的劣化分析方法相同的方法实施NEXAFS法。上述第三的劣化分析方法中,上述X射线的亮度、X射线的光子数、使用X射线进行扫描的能量范围,优选与上述第一的劣化分析方法的高亮度X射线的亮度、高亮度X射线的光子数、使用高亮度X射线进行扫描的能量范围相同。
NEXAFS由于可查看向非占有轨道的激发,很大程度上受到结合于考查元素的元素影响,因此认为能分离各个结合状态并能进行劣化因素的分离,NEXAFS可用于第三的本发明中的高分子劣化的分析。
具体而言,可通过与上述第一的劣化分析方法相同的方法进行实施。
通过使用上述电子产额法对高分子材料的X射线吸收光谱进行测定并分析,能分析高分子劣化度(%)。下面对其进行说明。
第三的本发明中,作为通过NEXAFS法求出硫交联的高分子材料中高分子劣化状态的手法,例如可举出如下方法:通过将上述X射线的能量为260~400eV的范围对碳原子K壳层吸收端的必要范围进行扫描,根据得到X射线吸收光谱,由下述(式3-1)计算出规格化常数α和β,对用该规格化常数α和β修正的碳原子K壳层吸收端的X射线吸收光谱进行波形分离,用得到的归属于285eV附近的π*跃迁的峰面积,由下述(式3-2)求出高分子劣化度(%)。
(式3-1)
[劣化前试样测定范围的X射线吸收光谱的总面积]×α=1
[劣化后试样测定范围的X射线吸收光谱的总面积]×β=1
(式3-2)
[1-[(劣化后π*的峰面积)×β]/[(劣化前π*的峰面积)×α]]×100=高分子劣化度(%)
由此,能得到劣化后的高分子(聚合物部分)的劣化程度(%),并能分析劣化率。此处,上述求出高分子劣化度的方法中,上述X射线的能量优选设定为260~350eV的范围。再者,上述求出劣化度的方法中,在进行上述(式3-1)的操作前,根据吸收端前的斜率进行评价并减去背景。
上述求出高分子劣化度的方法中,上述(式3-1)中X射线吸收光谱的总面积是对测定范围内的光谱进行积分得到的,并能根据测定条件等改变能量范围。
列举使用NR与BR的混合橡胶的新品、施行臭氧劣化7小时的试样(同时硫交联)的例子具体说明上述求出高分子劣化度的方法。
这些试样的碳原子K壳层吸收端的NEXAFS测定结果如图3-1所示。如图3-1所示的,劣化的试样虽然在285eV附近的πS峰与新品相比虽然变小,但难以用NEXAFS法测定绝对值。其理由是,从光源到试样的距离等微妙变化会对X射线吸收光谱的大小产生影响。根据以上理由,对于碳原子K壳层吸收端的NEXAFS的测定结果,不能进行试样间的简单比较。
因此,为了比较所测定的试样间的X射线吸收光谱,进行如下的规格化(为了能直接比较而修正各试样的X射线吸收光谱)。由于劣化前后碳壳层的X射线吸收量不变,因此用上述(式3-1),将碳原子K壳层吸收端的峰面积规格化为1。即,首先对规格化前的X射线吸收光谱,根据(式3-1)计算出规格化常数α、β,接着,通过对规格化前的X射线吸收光谱乘以α、β的光谱进行修正(规格化),能直接比较试样间的π*峰。
由此得到的规格化后的碳原子K壳层吸收端的光谱如图3-2所示。根据规格化的光谱,用上述(式3-2)确定高分子劣化度。上述高分子劣化度是从劣化前至劣化后的π*峰的减少率,表示试样中聚合物链的劣化率(%)。
再者,上述求出高分子劣化度的方法中,即使在上述(式3-2)中使用峰强度代替峰面积,也能同样求出高分子劣化度。
另外,上述中虽然对臭氧劣化品进行了说明,但对于氧劣化品、以臭氧与氧这两者方式进行劣化的劣化品,也能以同样的手法进行分析,能求出聚合物链的劣化度。
上述第三的本发明中高分子的劣化状态,例如,可使用佐贺县立九州同步加速器光研究中心的BL12束线进行分析。
而且,第三的本发明中,通过对硫交联的高分子材料照射一定能量的X射线,测定激发·释放的光电子,能求出硫交联的劣化状态。除了高分子的劣化状态之外,还可求出硫交联的劣化状态,由此能分析各自的劣化比例。
此处,作为对硫交联的高分子材料照射一定能量的X射线并测定激发·释放的光电子的手法,可举出X射线光电子分光法(XPS)等,具体而言,可通过使用通常的Al Kα1射线(1486.6eV)的XPS法、硬X射线光电子分光法(HAX-PES:Hard X-ray Photoemission Spectroscopy)等进行测定。
第三的本发明中,作为通过XPS法测定硫交联的劣化状态的手法,例如,可举出如下方法(方法1):对通过照射一定能量的X射线而激发·释放的光电子进行分光,对测定了对应于硫S2p的光电子强度的X射线光电子光谱进行波形分离,用得到的归属于硫氧化物的峰面积,由下述(式3-3)求出硫交联劣化度(%)。
(式3-3)
(归属于S2p的硫氧化物的峰面积)/(与S2p相关的总峰面积)×100=硫交联劣化度(%)
由此,能得到劣化后的硫交联部分的劣化度(%),并能分析劣化率。
上述方法1中,在上述(式3-3)中与S2p相关的总峰面积是对测定范围内的光谱进行积分得到的,并能根据测定条件等改变能量范围。
上述方法1中,从能测定与S2p(硫2p轨道)相关的峰面积的方面考虑,所使用的一定能量的X射线的能量范围优选为150~200eV,更优选为155~180eV。
列举使用NR与BR的混合橡胶的新品、实施热氧劣化1周的试样(同时硫交联)的例子具体说明上述方法1。
这些试样的高分子材料的S2p(硫2p轨道)的X射线光电子光谱的测定结果如图3-3所示。如图3-3所示的,可知劣化的试样中对应于S-S键的峰减少而对应于硫氧化物(SOX)的峰增加。因此,通过对劣化品的S2p的X射线光电子光谱波形分离为S-S键、SOX的各峰,将归属于SOX的峰面积与S2p相关的总峰面积适用于上述(式3-3),可求出硫交联劣化度(%)。
此处,虽然认为由于S2p轨道自旋轨道分裂,因此每个归属出现两个峰,另外硫氧化物(SOX)的峰是由价数不同的多个峰重叠而成的。但从原理上考虑,如图3-3所示的,可大体上分离为归属于S-S的峰和归属于SOx的峰。
再者,上述方法1中,即使在上述(式3-3)中使用峰强度代替峰面积,也能同样求出硫交联劣化度。
另外,作为通过HAX-PES法测定硫交联的劣化状态的方法,例如,可举出如下方法(方法2):对通过照射一定能量的X射线而激发·释放的光电子进行分光,对测定了对应于硫S1s的光电子强度的X射线光电子光谱进行波形分离,用得到的归属于硫氧化物的峰面积,由下述(式3-4)求出硫交联劣化度(%)。
(式3-4)
(归属于S1s的硫氧化物的峰面积)/(与S1s相关的总峰面积)×100=硫交联劣化度(%)
由此,能得到劣化后的硫交联部分的劣化度(%),能分析劣化率。
特别是通过使用HAX-PES法,其优点在于能测定通常的XPS法中不能测定的S1s轨道。即,用通常的XPS法虽然能测定S2p轨道的光谱,但由于所使用的X射线能量低,因此检测深度为表面~数nm。与此相对,用HAX-PES法能测定S1s轨道,由于所使用的X射线能量高,因此检测深度为表面~数十nm。因此,由于橡胶试样等硫交联高分子材料的最表面上产生硫化合物花斑,所以在测定最表面的XPS法中可能会影响测定结果,而HAX-PES法检测深度深,认为不受花斑的影响。因而,如果通过HAX-PES法,则特别是硫交联高分子材料的本体(内部)中硫交联的劣化分析成为可能。
再者,即使在用通常的XPS法测定S2p轨道的光谱的情况下,通过用氩离子刻蚀等去除最表面后进行测定,也能得到降低花斑影响的测定结果。
上述方法2中,在上述(式3-4)中对应于S1s的硫的总峰面积是对测定范围内的光谱进行积分得到的,并能根据测定条件等改变能量范围。
上述方法1中,从能测定与硫S1s(硫的1s轨道)相关的峰面积的方面考虑,所使用的一定能量的X射线的能量范围优选为2.5~15keV,更优选为4~10keV。
列举使用了NR与BR的混合橡胶的新品、实施热氧劣化1周的试样(同时硫交联)的例子具体说明上述方法2。
这些试样的高分子材料的S1s(硫的1s轨道)的X射线光电子光谱的测定结果如图3-4所示。如图3-4所示的,可知劣化的试样中对应于S-S键的峰减少而对应于硫氧化物(SOX)的峰增加。因此,通过对劣化品的S1s的X射线光电子光谱波形分离为S-S键、SOX的各峰,并将归属于SOX的峰面积与硫相关的总峰面积适用于上述(式3-4),求出硫交联劣化度(%)。
此处,虽然认为硫氧化物(SOX)的峰是价数不同的多个峰重叠而成的,但如果考虑到其原理,则如图3-4所示的,大体上能分离为归属于S-S的峰和归属于SOx的峰。
再者,上述方法2中,即使在上述(式3-4)中使用峰强度代替峰面积,也可同样求出硫交联劣化度。
另外,在上述方法1、2中虽然对氧劣化品进行了说明,但对臭氧劣化品、以臭氧与氧这两者方式进行劣化的劣化品也能以同样的手法进行分析,能求出硫交联的劣化度。
上述第三的本发明中的硫交联劣化状态,例如可使用Kratos制造的AXIS Ultra等通常的XPS装置、附带Spring-8BL46XU束线的HAX-PES装置进行分析。
而且,在第三的本发明的劣化分析方法中,例如可由下述(式3-5)计算出高分子劣化与硫交联劣化的贡献率。
(式3-5)
[高分子劣化度(%)]/[硫交联劣化度(%)]=高分子劣化与硫交联劣化的贡献率
即,通过使用上述的求出高分子劣化度的方法或求出硫交联劣化度的方法等来求出表示高分子和硫交联的各自劣化度的高分子劣化度(%)与硫交联劣化度(%)的比值(比例),能判断哪个劣化更进展。具体而言,上述(式3-5)中,高分子劣化与硫交联劣化的贡献率>1时,判断为高分子劣化的进展更大,而高分子劣化与硫交联劣化的贡献率﹤1时,判断为硫交联劣化的进展更大。因此,通过采用第三的本发明的方法,能制定与比以前更有效的耐劣化对策。
作为适用于第三的本发明的上述硫交联高分子材料,并没有特别限定,可举出以前公知的材料,例如,适宜使用含有一种以上二烯系橡胶的硫交联橡胶材料,该橡胶材料与一种以上的树脂进行复合而得到的硫交联复合材料。作为上述二烯系橡胶、树脂,可使用与上述第一的本发明相同的材料等。第三的本发明的劣化分析方法可适用于这些材料。
实施例
根据实施例对本发明进行具体说明,但本发明并不局限于这些实施例。
(实施例1-1~1-5和比较例1-1)
用于实施例和比较例的劣化后的试样是通过以下的橡胶材料和劣化条件制作的。再者,为了以NEXAFS法进行测定,通过薄切片机将试样加工成100μm以下的厚度。然后,为了避免劣化以外的氧的影响出现,试样制作后保存于真空干燥器中。
(橡胶材料)
IR:ニッポールIR 2200 日本瑞翁株式会社制造
BR:ウベポールBR 130B 宇部兴产株式会社制造
SBR:ニッポール1502 日本瑞翁株式会社制造
NR:TSR20 海南中化橡胶有限公司制造
北美行驶品:在北美已经行驶过的轮胎(NR与BR的混合橡胶)
中近东行驶品:在中近东已经行驶过的轮胎(NR与BR的混合橡胶)
(劣化条件)
臭氧劣化:40℃ 50pphm(1小时)
氧劣化:80℃ 空气中(7天)
(使用装置)
NEXAFS:佐贺县立九州同步加速器光研究中心的附带BL12光束线的NEXAFS测定装置
XPS:Kratos公司制造AXIS Ultra
通过使用NEXAFS,对各试样实施以下的劣化率分析,测定劣化度(%)。
再者,NEXAFS的测定条件如下所述。
亮度:5×1012光子/s/mrad2/mm2/0.1%bw
光子数:2×109光子/秒
(劣化度分析)
将高亮度X射线的能量为260~400eV的范围进行扫描,得到碳原子K壳层吸收端的X射线吸收光谱。根据该光谱中必要范围为260~350eV的范围,由(式1-1)计算出规格化常数α、β,使用该常数对光谱进行规格化(修正)。使用Gauss函数,对规格化后的光谱中归属于285eV附近的π*跃迁的峰波形分离为各聚合物成分的归属峰。根据所归属的各峰面积,由(式1-2)求出各聚合物成分的劣化度(%)。
关于比较例1-1,使用XPS对劣化后的试样进行评价。
根据以上分析得到的结果如表1所示。
[表1]
使用XPS的比较例1-1中,不能对混合橡胶中所含的各二烯系聚合物的劣化度进行分离并分析。与此相对,使用NEXAFS的实施例1-1~1-5中,通过对π*跃迁峰波形分离为各二烯系聚合物,能对各聚合物成分进行劣化度分析,证明了第一的本发明的评价法的有效性。因此,能期待第一的本发明适用于轮胎等含有两种以上二烯系聚合物的高分子材料的耐劣化对策等。
(实施例2-1~2-2和比较例2-1~2-2)
用于实施例和比较例的劣化后的试样是通过以下的橡胶材料和劣化条件制作的。再者,为了以XPEEM和TEM进行测定,通过薄切片机将试样加工成100μm以下的厚度。然后,为了避免劣化以外的氧的影响出现,试样制作后保存于真空干燥器中。
(橡胶材料)
IR:ニッポールIR 2200 日本瑞翁株式会社制造
NR:TSR20 海南中化橡胶有限公司制造
SBR:SBR1502 LG化学品公司制造
北美行驶品:在北美已经行驶过的轮胎(NR与SBR的混合橡胶)
(劣化条件)
臭氧劣化:40℃ 50pphm(1小时)
(使用装置)
XPEEM:SPring-8 附带BL17SU束线的分光型光电子/低能量电子显微镜(SPELEEM:Elmitec公司制造)
TEM:日本电子株式会社制造的JEM2100F
通过使用XPEEM,对各试样实施以下的劣化率分析,测定劣化度(%)。
再者,XPEEM的测定条件如下所述。
光子数:1×1011光子/秒
(各聚合物的观察)
对各试样,如能对混合的每个聚合物进行外观观察,则以○表示。
(劣化度分析)
将高亮度X射线的能量为260~400eV的范围进行扫描,得到碳原子K壳层吸收端的各聚合物成分的X射线吸收光谱。根据该光谱中必要范围为260~350eV的范围,由(式2-1)计算出各聚合物成分的规格化常数α、β,使用该常数对光谱进行规格化(修正)。对规格化后的光谱进行波形分离,根据归属于285eV附近的π*跃迁的峰面积,由(式2-2)求出各聚合物成分的劣化度(%)。
(劣化贡献率分析)
将高亮度X射线的能量为500~600eV的范围进行扫描,得到氧原子K壳层吸收端的各聚合物成分的X射线吸收光谱。对该光谱进行波形分离,将峰顶为532eV以上且小于532.7eV的低能量一侧的峰作为氧劣化,峰顶为532.7eV以上且534eV以下的高能量一侧的峰作为臭氧劣化,由(式2-3)计算出各聚合物成分的氧劣化与臭氧劣化的贡献率。
(劣化指标测定)
根据上述劣化率分析得到的劣化后的碳原子K壳层吸收端的X射线吸收光谱,由(式2-4)求出各聚合物成分的规格化常数γ。使用该常数,由(式2-5)对氧原子K壳层吸收端的总面积进行修正(规格化),求出氧和臭氧在各聚合物成分中的结合量(劣化指标)。
关于比较例2-1和2-2,使用TEM对劣化后的试样进行评价。
根据以上分析得到的结果如表2所示。
[表2]
使用TEM的比较例2-1和2-2中,不能对混合橡胶中所含的各二烯系聚合物的劣化度、氧劣化贡献率、臭氧劣化贡献率以及氧和臭氧的结合量进行分离并分析。与此相对,使用XPEEM法的实施例2-1和2-2中,通过对混合橡胶中的每个二烯系聚合物进行X射线吸收光谱的测定,能对它们进行分离并分析,证明了第二的本发明的评价法的有效性。
因此,能期待第二的本发明适用于轮胎等含有两种以上二烯系聚合物的高分子材料的耐劣化对策等。
(实施例3-1~3-8和比较例3-1~3-4)
用于实施例和比较例的劣化后的试样是通过以下的橡胶材料和劣化条件制作的。再者,在用NEXAFS法进行测定时,通过薄切片机将试样加工成100μm以下的厚度,然后,为了避免劣化以外的氧的影响出现,试样制作后保存于真空干燥器中。
(橡胶材料)
NR与BR的混合橡胶(硫交联):TSR20 海南中化橡胶有限公司制造,ウベポールBR 130B 宇部兴产株式会社制造
中近东行驶品:在中近东已经行驶过的轮胎(NR与BR的混合橡胶(硫交联),使用侧壁)
(劣化条件)
臭氧劣化:40℃50pphm
氧劣化:80℃空气中
(使用装置)
NEXAFS:佐贺县立九州同步加速器光研究中心的附带BL12束线的NEXAFS测定装置
XPS:Kratos公司制造AXIS Ultra
HAX-PES:SPring-8 附带BL46XU束线的HAX-PES装置
通过使用NEXAFS,对劣化前后的各试样实施以下的分析,测定高分子劣化度(%)。
再者,NEXAFS的测定条件如下所述。
亮度:5×1012光子/s/mrad2/mm2/0.1%bw
光子数:2×109光子/秒
(高分子劣化度分析)
将X射线的能量为260~400eV的范围进行扫描,得到碳原子K壳层吸收端的X射线吸收光谱。根据该光谱中必要范围为260~350eV的范围,由(式3-1)计算出规格化常数α、β,使用该常数对光谱进行规格化(修正)。对规格化后的光谱进行波形分离,根据归属于285eV附近的π*跃迁的峰面积,由(式3-2)求出高分子劣化度(%)。
通过使用XPS,对劣化后的各试样实施如下方法1的分析,测定硫交联劣化度(%)。
再者,XPS的测定条件如下所述。
测定光源:Al(单色器)
照射X射线的能量:1486eV
测定输出:20kV×10mA
测定元素和轨道:S2p
束缚能量:163.6eV(S2p1/2)、162.5eV(S2p3/2)
(硫交联劣化度分析(方法1))
对通过照射上述一定能量的X射线而激发·释放的光电子进行分光,得到测定了对应于硫S2p的光电子强度的X射线光电子光谱。对该光谱160~175eV的范围分别进行波形分离为对应于164eV附近的S-S键和168eV附近的SOx的峰,用得到的归属于硫氧化物的峰面积与160~175eV范围的硫的总峰面积,由(式3-3)求出硫交联劣化度(%)。
通过使用HAX-PES,对劣化后的各试样实施如下方法2的分析,测定硫交联劣化度(%)。
再者,HAX-PES的测定条件如下所述。
测定光源:高亮度X射线
照射X射线的能量:8keV
测定输出:1013光子/秒
测定元素和轨道:S1s
束缚能量:2472eV
(硫交联劣化度分析(方法2))
对通过照射上述一定能量的X射线而激发·释放的光电子进行分光,得到测定了对应于硫S1s的光电子强度测定的X射线光电子光谱。对该光谱2465~2480eV的范围分别进行波形分离为对应于2470eV附近的S-S键和2472eV附近的SOx的峰,用得到的归属于硫氧化物的峰面积与2465~2480eV范围的硫的总峰面积,由(式3-4)求出硫交联劣化度(%)。
(高分子劣化与硫交联劣化的贡献率分析)
将上述高分子劣化度分析和硫交联劣化度分析(方法1-2)中所求出的高分子劣化度和硫交联劣化度的值适用于(式3-5),计算出高分子劣化与硫交联劣化的贡献率。
上述高分子劣化度分析、硫交联劣化度分析(方法1)中得到的结果如表3-1所示,上述高分子劣化度分析、硫交联劣化度分析(方法2)中得到的结果如表3-2所示。
[表3-1]
[表3-2]
使用Swell或FT-IR的比较例3-1~3-4中,都不能对劣化后的试样的高分子劣化度、硫交联劣化度、贡献率进行分析。与此相对,使用NEXAFS和XPS的实施例3-1~3-4、使用NEXAFS和HAX-PES的实施例3-5~3-8中,每种分析均可进行,证明了第三的本发明的评价法的有效性。特别是使用HAX-PES时,认为不受试样表面的花斑影响,因此推测实施例3-5~3-8的结果是信赖性更高的结果。
- 还没有人留言评论。精彩留言会获得点赞!