一种36Cl与41Ca相结合的核素定年法的制作方法
本发明涉及一种36Cl与41Ca相结合的核素定年法,属于定年技术领域。
背景技术:
地球表面的形状与演化历史是地形学的基本研究内容,为了理解地形的演化和大陆的剥蚀,人们不仅需要了解与之有密切关系的物理过程,同时也有必要知道这些地形特征的形成时间及改变快慢。目前,常见的测年方法有14C测年法、10Be测年法和26Al测年法等,各有利弊。其中,14C测年法是选择放射性碳核素及其衰变后的同位素为研究对象,根据14C衰变来计算出样品的大概年代的一种测年方法。目前它主要应用于考古学和第四纪地质研究,例如青藏高原盐湖演化研究、河湖演化研究、洞穴堆积物测年、地下水研究、海陆变迁与海面变化研究等。但是,14C测年法也只能准确测出5、6万年以内的地质构造及出土文物,对于年代更久远的事件,如生活在五十万年以前的周口店北京猿人,实际上利用14C测年法是无法测定出来的。10Be测年法和26Al测年法的测年范围在几百万年以上,测量年限过长,对于地质构造断代有局限性,而且该法需要的样品量较大,为了构建等时线,需依据具体情况采集分析多个样品,对数据精度的要求越高,所需的样品数量就越多。此外,该法一般只适用于同一层位,相近探方可获得多个石英质砾石的地点。
36Cl测年法是选择36Cl核素及其衰变后的同位素为研究对象,根据36Cl衰变的程度来计算出样品的大概年代的一种测年方法,其测年原理与上面所讲的14C测年法相似,它们同属于放射性同位素测年法。36Cl测年法是从20世纪70年代末期随着AMS和36Cl样品制备技术的发展从而逐渐发展起来。36Cl的半衰期为0.305Ma,测年范围最大可达3.05Ma(10倍半衰期),所以36Cl测年法可以填补许多年轻沉积地层无法确定年龄的空白,对第四纪地层年代测定具有重要作用。如果单用36Cl,必须要侵蚀速率数据,所以也限制了36Cl测年法的使用。
41Ca是钙的放射性同位素之一,它的半衰期是(1.03±0.04)×105年,可以用来测定104-106a地质样品的年龄。与36Cl结合能很好的消除侵蚀速率对结果的影响,在计算结果的处理上能够给出可靠数据。
AMS加速器质谱的工作原理:利用加速器把从离子源引出的被测核素的带电离子及其干扰离子加速到一定的能量,再利用磁分析器和静电分析器将不同质量数的核素离子分开,进行核素分析。一方面利用加速器可以把粒子加速到较高的能量(几MeV至几百MeV),从而通过电子剥离介质使离子处于较高的电荷态(q≥3),使分子离子离解,以消除分子离子的干扰;另一方面采用重离子探测器,可以对同量异位素进行鉴别,消除同量异位素的干扰。AMS的优点:(1)测量灵敏度高;(2)样品用量少;(3)测量时间短。
在目前已有测年方法中,14C测年法和Be-Al测年法因半衰期限制,两者在几十万年尺度范围内断代精度较差。本发明应用36Cl与41Ca相结合的综合测年法,正好填补了上述空缺,对研究第四纪中更新世至晚更新世中期气候演变和考古工作有重大意义。本发明通过测量36Cl/Cl和41Ca/40Ca的比值和含量确定岩石暴露和埋藏年龄,克服侵蚀速率的影响。在测量计算中,两者综合运用比用单一核素测年,更为精确,大大提高了测年的精确度。
技术实现要素:
本发明的目的是解决现有技术的不足,提供一种36Cl与41Ca相结合的核素定年法。本发明能够解决现有技术中测年方法精确度不够、测年范围过窄等问题,可以准确地测量出地质岩石的暴露和埋藏年龄。
本发明解决上述技术问题的技术方案如下:一种36Cl与41Ca相结合的核素定年法,包括如下步骤:
(1)样品的采集
选择体积≥1.5m×2.5m×2m、方解石含量≥75%的石灰岩作为取样点,取样深度距离地表≤10cm,得到样品;
(2)样品的表面去污
2a.将步骤(1)得到的样品,粉碎至0.5~1mm的粒径后,放入锥形瓶中,用足量超纯水充分振荡后,去掉上层的漂浮物,用超纯水漂洗、去除浮在上层的颗粒≥5次,再向锥形瓶中加入适量超纯水,超声清洗10min,重复超声清洗≥3次,得到混合液A;
2b.在步骤2a得到的混合液A,缓慢加入适量8M的稀硝酸,立即振荡至样品完全溶解,然后盖上蒸发皿,静置30~60min直到不再有气泡冒出,得到混合液B;
2c.将步骤2b得到的混合液B,超声清洗10min,倒掉上层悬浊液,用超纯水清洗≥5次;
2d.重复上述步骤2b和2c,得到混合液C;
2e.将步骤2d得到的混合液C,超声清洗10min,倒掉上层悬浊液,用超纯水清洗≥5次,得到混合液D;
2f.将步骤2e得到的混合液D,静置,倒掉上层清液,于110℃烘干2h后,称重,并存放于聚丙烯塑料瓶中,得到表面去污后的样品;
(3)Cl的化学制备
3a.在步骤(2)得到的表面去污后的样品中,缓慢加入4M的稀硝酸,并保持反应溶液的pH 2~3,待上述表面去污后的样品充分溶解后,过滤,收集过滤液,得到过滤液A;
3b.在步骤3a收集到的过滤液A中,加入过量的0.25M的AgNO3溶液,加热1~2h后,黑暗中放置24h,得到过滤液B;
3c.将步骤3b得到的过滤液B,离心,去掉上层清液,用去离子水振荡冲洗白色沉淀物≥5次后,再离心,去掉上层清液,得到白色沉淀物A;
3d.在步骤3c得到的白色沉淀物A中,加入适量的去离子水,再加入纯度为99.999%的高纯氨水至过量,保持pH值>10,充分搅拌,至上述白色沉淀物A全部溶解,得到溶液C;
3e.在步骤3d得到的溶液C中,加入饱和的Ba(NO3)2溶液,摇匀,温水静置过夜,保持反应溶液的pH>10,得到溶液D;
3f.将步骤3e得到的溶液D,过滤,去除沉淀,滤液保留;
3g.在步骤3f得到的滤液中,加入过量的纯度为65%的高纯浓硝酸溶液,保持反应溶液的pH<2,得到含有白色沉淀物B的溶液E,过滤得到白色沉淀物B;
3h.将步骤3g得到的白色沉淀物B,离心,沉淀保留,再用纯度为99.9%的优级纯的乙醇和4M的稀硝酸漂洗上述沉淀物,得到沉淀物C;
3i.将步骤3h得到的沉淀物C用铝箔密封,于100℃放置≥8h,称重,储存于棕色玻璃瓶中,即为化学制备得到的Cl样品;
(4)Ca的化学制备
4a.将步骤(2)得到的表面去污后的样品研成粉末,离心管中加入6mol/L的HCl溶液,充分混匀后静置1h,3600r/min离心10min,将上清液移至另一离心管中;
4b.向离心管缓慢加入氨水至pH值为12.4,静置1h,3600r/min离心10min,将上清液移至另一离心管中;
4c.向步骤4b得到的上清液中加入HNO3溶液酸化,加去离子水稀释至15mL,过阳离子交换玻璃柱,流速控制在1-1.5mL/min,先用15mL 0.8mol/L的HNO3洗脱41K,再用4mol/L的HNO3洗脱Ca2+,收集流出液于离心管中,加去离子水,制备成纯化的Ca2+预处理液;
4d.二次氟化法制备CaF2:在步骤4c得到的纯化的Ca2+预处理液中加入优级纯的HF溶液,生成白色不溶物,隔夜放置,3600r/min离心10min,得到CaF2沉淀,用去离子水洗涤上述CaF2沉淀两次,进行一次氟化反应,100℃烘烤24h后置于干燥Ar气中保存,得到一次氟化后的CaF2;称取50mg的上述一次氟化后的CaF2,与200mg的NH4HF2均匀混合,进行二次氟化反应,称重后于干燥Ar气环境中保存,即得到二次氟化法制备的CaF2;
(5)样品中Cl及Ca的测量
将已知同位素丰度的标准样品、步骤(3)得到的化学制备的Cl样品、步骤(4)得到的化学制备的Ca样品,按照41Ca/40Ca和36Cl/35Cl同位素丰度从低到高的顺序,依次压到铜靶锥中,然后在铜质靶锥末端垫上纯度为99.9%的高纯银粉,接着采用加速器质谱方法测量,得到样品中41Ca/40Ca和36Cl/35Cl比值;
(6)结果分析
将步骤(5)得到的样品中的41Ca/40Ca和36Cl/35Cl比值,与已知同位素丰度的标准样品的41Ca/40Ca和36Cl/35Cl比值进行对比,即可得到被测样品中41Ca/40Ca和36Cl/35Cl比值的真实数值,再在已知样品中稳定同位素量的情况下,通过测量样品中的41Ca/40Ca和36Cl/35Cl原子数含量值,即可得到样品的41Ca和36Cl的含量,即得到样品的核素定年法数值。
本发明的步骤4a中,HCl为分析纯,北京化学试剂研究所;将上清液移至另一离心管中,是为了除去不溶于HCl溶液的SiO2和其它化合物。
步骤4c中,HNO3为分析纯,北京化学试剂研究所;加HNO3酸化的作用是:增加环境的氧化性,因为在步骤4d中HF溶液是弱酸,普通环境下氟化不完全,增加环境的氧化性有助于提高氟化程度。
步骤4d中,HF为分析纯,北京化学试剂研究所。
在上述技术方案的基础上,本发明还可以做如下改进。
进一步,步骤(1)所述取样位置为山脚、山腰和山顶,采样的同时记录样品的经纬度、海拔及采样点附近的地表覆盖情况。
进一步,步骤(2)中所述超声的频率≥20KHz,功率密度为0.3w/cm2。
进一步,步骤4b所述pH值为12.4时,Fe(OH)3、Al(OH)3、Mg(OH)2沉淀完全析出。
更进一步,所述Fe(OH)3完全析出时pH值为4.1,所述Al(OH)3完全析出时pH值为5.2,所述Mg(OH)2完全析出时pH值为12.4。
进一步,步骤4c所述阳离子交换玻璃柱的型号为001×8强酸性阳离子交换树脂,内径6cm,树脂粒径为100-200目,树脂层高8cm。
进一步,步骤4d所述一次氟化和二次氟化均在密封管式炉中进行。
本发明的有益效果是:
(1)在目前已有测年方法中,14C测年法和Be-Al测年法因半衰期限制,均缺少中间精度(105年),而本发明的36Cl与41Ca相结合的核素定年法正好填补了这个空缺,对研究第四纪中更新世至晚更新世中期气候演变和考古工作有重大意义。
(2)在测量计算中,本发明通过测量41Ca/40Ca和36Cl/35Cl的比值得到41Ca和36Cl的含量,相比用单一核素测年法,侵蚀速率更为精确,大大提高了测年的精确度。
(3)本发明所采用的41Ca和36Cl这两种核素测量都能用AMS实现,对样本的测量可同步进行,操作简便,工作效率高。
(4)36Cl半衰期为3×105年,41Ca半衰期为1×105年,两者的测年范围相差适中,能很好的消除侵蚀速率对结果的影响,在计算结果的处理上能够给出可靠数据。
(5)本发明的方法简单,市场前景广阔,适合规模化应用。
附图说明
图1为本发明的埋藏等时线图(x1:T=180000a,x2:T=130000a,x3:T=80000a,x4:T=30000a,x5:T=20000a)
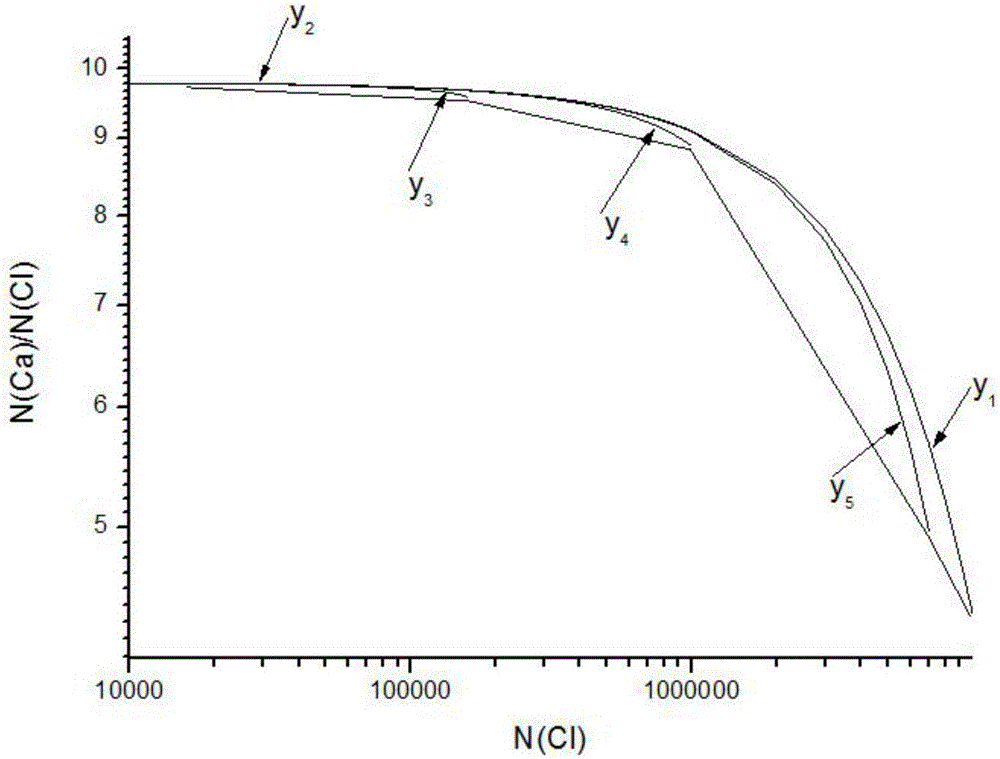
图2为本发明的侵蚀速率曲线图(y1:侵蚀速率为0cm·a-1,y2:侵蚀速率为0.1cm·a-1,y3:侵蚀速率为0.01cm·a-1,y4:侵蚀速率为0.001cm·a-1,y5:侵蚀速率为0.0001cm·a-1)
图3为本发明以暴露时间t为常数绘制的埋藏等时线图(z1:t=1500000a,z2:t=1600000a,z3:t=2000000a,z4:t=15000000a,z5:t=652621.5983a)。
图4是将图1、图2、图3结合,生成一幅通过测量沉积地层某些矿物中就地产生的36Cl/41Ca比值和36Cl的浓度,带入其中就可以获得该地层的暴露年龄及埋藏年龄。
图5为本发明的41Ca的制取流程图。
图6为本发明的36Cl的制取流程图。
图7为本发明的41Ca/36Cl和36Cl的暴露与埋藏曲线的天坑测量点-A点。
图8为本发明实施例中空白样品的36Cl测量图谱(横坐标表示离子总能量道数,纵坐标表示粒子在探测器E1极板上收集的能量道数)。
图9为本发明实施例中岩石样品的36Cl测量图谱(横坐标表示离子总能量道数,纵坐标表示粒子在探测器E1极板上收集的能量道数)。
图10为本发明实施例中空白样品的41Ca测量图谱(横坐标表示离子总能量道数,纵坐标表示粒子在探测器E1极板上收集的能量道数)。
图11为本发明实施例中岩石样品的41Ca测量图谱(横坐标表示离子总能量道数,纵坐标表示粒子在探测器E1极板上收集的能量道数)。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
一种36Cl与41Ca相结合的核素定年法,包括如下步骤:
(1)样品的采集
在广西天坑选择体积≥1.5m×2.5m×2m、方解石含量≥75%的石灰岩作为取样点,取样深度距离地表≤10cm,得到样品。
(2)样品的表面去污
2a.将步骤(1)得到的样品,粉碎至0.5~1mm的粒径后,放入锥形瓶中,用足量超纯水充分振荡后,去掉上层的漂浮物,再向锥形瓶中加入适量超纯水,超声清洗10min,用超纯水漂洗、去除浮在上层的颗粒≥5次,重复超声清洗≥3次,得到混合液A;
2b.在步骤2a得到的混合液A,缓慢加入适量8M的稀硝酸,立即振荡至样品完全溶解,然后盖上蒸发皿,静置30~60min直到不再有气泡冒出,得到混合液B;
2c.将步骤2b得到的混合液B,超声清洗10min,倒掉上层悬浊液,用超纯水清洗≥5次;
2d.重复上述步骤2b和2c,直到步骤(1)得到的样品的表面去除率≥10%,得到混合液C;
2e.将步骤2d得到的混合液C,超声清洗10min,倒掉上层悬浊液,用超纯水清洗≥5次,得到混合液D;
2f.将步骤2e得到的混合液D,静置,倒掉上层清液,于110℃烘干2h后,称重,并存放于聚丙烯塑料瓶中,得到表面去污后的样品;
2g.所需岩石样品的量取决于岩石中Cl的浓度、36Cl的浓度,样品中Cl含量越高,单位质量岩石样品得到的AgCl越多,引束时间越长;样品中36Cl含量越高,相同引出束流、相同传输效率情况下电离室得到的计数率越高,达到相同统计误差所需的时间越短;
设岩石样品质量为M/g,36Cl的浓度为M36Cl/atom·g-1,加速器离子源引出效率为η,总粒子传输效率为ε,则在管道终端的多阳极电离室中得到的计数率为n,实验要求达到的统计误差为ν,则有:
ν2·n·t=1
M·M36Cl·ε·η=n·t
上两式联立可得:
实验要求的统计误差、以往实验的传输效率以及样品36Cl含量的初步估计可以计算出需要的原始样品最小量,由于实验中不可能将样品全部引出,而且离子源引出效率不可能为100%(实际为10-2的量级),实际取样时应当比上述计算量大100倍左右,以保证达到预期的实验效果(即具体岩石取样量为100g左右)。
(3)Cl的化学制备,制取流程如图5所示:
3a.在步骤(2)得到的表面去污后的样品中,缓慢加入4M的稀硝酸,并保持反应溶液的pH 2~3,待上述表面去污后的样品充分溶解后,过滤,收集过滤液,得到过滤液A;
3b.在步骤3a收集到的过滤液A中,加入过量的0.25M的AgNO3,加热1~2h后,黑暗中放置24h,得到过滤液B;
3c.将步骤3b得到的过滤液B,离心,去掉上层清液,用去离子水振荡冲洗白色沉淀物≥5次后,再离心,去掉上层清液,得到白色沉淀物A;
3d.在步骤3c得到的白色沉淀物A中,加入适量的去离子水,再加入纯度为99.999%的高纯氨水至过量,保持pH值>10,充分搅拌,至上述白色沉淀物A全部溶解,得到溶液C;
3e.在步骤3d得到的溶液C中,加入饱和的Ba(NO3)2溶液,摇匀,温水静置过夜,保持反应溶液的pH>10,得到溶液D;
3f.将步骤3e得到的溶液D,过滤,去除沉淀,滤液保留;
3g.在步骤3f得到的滤液中,加入过量的纯度为65%的高纯浓硝酸溶液,保持反应溶液的pH<2,得到含有白色沉淀物B的溶液E,过滤得到白色沉淀物B;
3h.将步骤3g得到的白色沉淀物B,离心,沉淀保留,再用纯度为99.9%的优级纯的乙醇和4M的稀硝酸漂洗上述沉淀物,得到沉淀物C;
3i.将步骤3h得到的沉淀物C用铝箔密封,于100℃放置≥8h,称重,储存于棕色玻璃瓶中,即为化学制备得到的Cl样品。
(4)Ca的化学制备,制取流程如图6所示:
4a.在步骤(2)得到的表面去污后的样品用玛瑙研钵将石灰岩样品研成粉末,离心管中加入1L6mol/L的HCl溶液(分析纯,北京化学试剂研究所),充分混匀后静置1h,3600r/min离心10min,将上清液移至另一离心管中(以除去不溶于HCl溶液的SiO2和其它化合物);
4b.向离心管缓慢加入氨水(优级纯,北京化学试剂研究所)至pH值为12.4,此时Fe(OH)3、Al(OH)3、Mg(OH)2沉淀完全析出(其中,Fe(OH)3完全析出时pH值为4.1,Al(OH)3完全析出时pH值为5.2,Mg(OH)2完全析出时pH值为12.4,20℃时饱和Ca(OH)2水溶液的pH值为12.65,通常认为离子浓度小于10-5即为完全沉淀),静置1h,3600r/min离心10min,将上清液移至另一离心管中;
4c.向该上清液中加入1L HNO3溶液(分析纯,北京化学试剂研究所,下同)酸化(加HNO3的作用还有增加环境的氧化性,因为HF溶液是弱酸,普通环境下氟化不完全,增加环境的氧化性有助于提高氟化程度),加去离子水稀释至15mL,过001×8强酸性阳离子交换玻璃柱(内径6cm,树脂粒径为100-200目,树脂层高8cm),流速控制在1-1.5mL/min,先用15mL 0.8mol/L的HNO3洗脱41K,再用4mol/L的HNO3洗脱Ca2+,收集流出液于离心管中,加去离子水,制备成纯化的Ca2+预处理液;
4d.二次氟化法制备CaF2:在步骤4c得到的纯化Ca2+预处理液中加入优级纯的HF溶液(北京化学试剂研究所),生成白色不溶物,隔夜放置,3600r/min离心10min,用去离子水洗涤CaF2沉淀两次,在密封管式炉中进行一次氟化反应,100℃烘烤24h后置于干燥Ar气中保存,得到一次氟化后的CaF2;称取50mg的上述一次氟化后的CaF2,与200mg的NH4HF2均匀混合,在密封管式炉中进行二次氟化反应,称重后于干燥Ar气环境中保存,即得到二次氟化法制备的CaF2。
(5)样品中Cl及Ca的测量
将已知同位素丰度的标准样品、步骤(3)得到的化学制备的Cl样品、步骤(4)得到的化学制备的Ca样品,按照41Ca/40Ca和36Cl/35Cl同位素丰度从低到高的顺序,依次压到铜靶锥中,然后在铜质靶锥末端垫上纯度为99.9%的高纯银粉,压靶完毕后,压好样品的靶锥要避光单独保存在小离心管中,接着采用加速器质谱系统,得到样品的41Ca和36Cl含量,包括如下步骤:
5a.束流传输、传输状态的确定:主要包括加速器测量中37Cl和41Ca的束流传输、36Cl和41Ca的模拟传输以及最终传输条件的确定;
5b.对△E-Q3D磁谱仪以及多阳极气体探测器进行参数设置;
5c.△E-Q3D系统以及多阳极探测器对同量异位素36S/36Cl和41K/41Ca的鉴别;
通过束流归一以及扣除本底干扰可得出,天然岩石样品中41Ca/40Ca的本底水平<8×10-14。
(6)结果分析
将步骤(5)得到的样品的41Ca/40Ca(41Ca/40Ca=5.1×10-13)和36Cl/35Cl(36Cl/35Cl=3.41×10-12)值,与标准样品的41Ca/40Ca(41Ca/40Ca=3×10-11)和36Cl/35Cl(36Cl/35Cl=4.47×10-11)值进行对比,即可得到被测样品中41Ca/40Ca和36Cl/35Cl值的真实数值,再在已知同位素丰度的标准样品中稳定同位素量的情况下,通过测量样品中的41Ca和36Cl原子数含量值,即可得到样品的41Ca(4.9×106atoms/g)和36Cl(5.3×105atoms/g)的含量,即得到样品的核素定年法。根据实验结果,对天坑暴露年龄进行了计算,最终给出了天坑在不考虑侵蚀速率的情况下的暴露年龄:22±9ka。
图7中,A点为实际测量点。
图9中,可以直接从图中读出36Cl的含量为5.3×105atoms/g。
图11中,可以直接从图中读出41Ca的含量为4.9×106atoms/g。
结论:
由图1可知,36Cl的含量值相同,41Ca/36Cl的含量值随着埋藏年龄T的增大而减少;41Ca/36Cl的含量值相同,36Cl的含量值随着埋藏年龄T的增大而减少。
由图2可知,侵蚀速率越大,达到极限浓度(平衡浓度)所需的时间越短,反之侵蚀速率越小,样品中36Cl浓度达到极限(平衡浓度)所需的时间越长;在没有侵蚀速率时,达到极限浓度的时间为无限长。
由图3可知,暴露时间相同,41Ca/36Cl的N值随36Cl的N值增大而增大;暴露时间越大,相应的36Cl及41Ca/36Cl的N值也越大。
图4由图1、2、3总结而来。
对比试验
取深度为10m的深层岩石,作为空白样品,其41Ca/40Ca的含量低于8×10-14,36Cl/35Cl的含量范围在(1.48±1.45)×10-14。从图8、图10中可以看出内层样品中的36Cl、41Ca几乎没有,所以可以用岩石的表面样品来测量暴露年龄。由此可见,本发明的方法,两种核素都能用AMS实现,对样本的测量可同步进行,操作简便,工作效率高,测年精确度高,简单可行,可以准确地测量出地质岩石的暴露和埋藏年龄,对研究第四纪中更新世至晚更新世中期气候演变和考古工作有重大意义。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!