同时测定独一味药材中8种成分的UPLC方法与流程
本发明涉及药物质量控制方法
技术领域:
,具体涉及同时测定独一味药材中8种成分的UPLC方法。
背景技术:
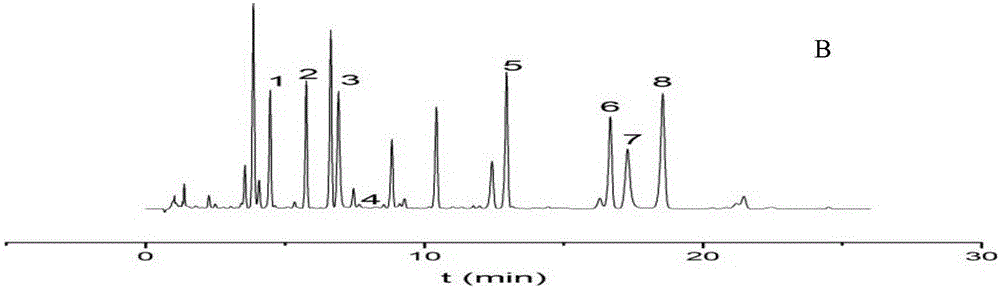
:独一味(Lamiophlomisrotata(Benth.)Kudo)系藏族习用药材,为唇形科植物,主要分布于我国西藏、云南、四川、青海、甘肃等地。药用部位为独一味的干燥地上部分,生长于海拔2700-4500米的高原或高山上强度风化的碎石滩中或石质高山草甸、河滩地(四川植物志、云南植物志),主要成分有黄酮类、环烯醚萜类和苯乙醇苷类。目前,测定独一味药材中各种成分方法通常都是HPLC法。然而,HPLC法检测的时间较长,效率不高。同时,从数量上看,其最多同时检测独一味药材中的7种成分,而考虑到中药材的活性成分繁多,要更加可靠、更加全面地监控独一味药材的质量,也有必要对检测成分的种类进行扩展。技术实现要素:为解决上述问题,本发明提供了一种同时测定独一味药材中8种成分的UPLC方法,其特征在于:所述8种成分为胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷和麦角甾苷,包括以下步骤:(1)8种成分标准曲线的建立:a、对照品溶液的制备:取胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷、麦角甾苷对照品,混合,加甲醇配制成混合对照品溶液;b、对照品溶液的测定:配制系列浓度的混合对照品溶液,分别注入UPLC色谱仪,梯度洗脱,测定各色谱峰峰面积,得到胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷和麦角甾苷的标准曲线;色谱条件如下:检测波长:238nm、320nm和350nm;色谱柱:C18色谱柱;流动相:以水或0.1%磷酸水溶液为流动相A,乙腈为流动相B;梯度洗脱:0~10min,6%~14.5%B;10~18.5min,14.5%~17%B;18.5~23min,17%~18%B;(2)待测样品中8种成分的含量测定:c、供试品溶液的制备:取待测独一味药材粉末,加甲醇提取,过滤得供试品溶液;d、供试品溶液的测定:取供试品溶液,注入UPLC色谱仪,以步骤b相同的色谱条件检测,根据步骤(1)的标准曲线得到待测样品中胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷和麦角甾苷的含量。进一步地,步骤c中,所述独一味药材粉末的粒径为过4号筛。进一步地,步骤c中,所述甲醇为70%甲醇。进一步地,所述甲醇与独一味药材的体积质量比为60mL:1g。进一步地,步骤c中,所述提取为回流提取。进一步地,回流提取的温度为80℃。进一步地,所述回流提取的时间为60min。进一步地,所述过滤为0.22μm微孔滤膜过滤。进一步地,所述色谱柱为Kinetex2.6μmXB-C18色谱柱。进一步地,所述色谱条件的柱温为30℃。进一步地,所述色谱条件的流速为1.0mL/min。本发明成功建立了同时测定独一味药材中8种成分的UPLC方法,该方法准确可靠、简便快速。本发明的方法,为全面监控独一味药材质量提供了有效的保障。显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。附图说明图1和图2分别为对照品(A)及独一味地上部分(B)色谱图;各色谱峰的归属如下1.胡麻属苷;2.山栀苷甲酯;3.绿原酸;4.咖啡酸;5.8-O-乙酰山栀苷甲酯;6.连翘酯苷B;7.木犀草苷;8.麦角甾苷。图3为提取方法筛选的结果。图4为提取时间筛选的结果。图5为提取溶剂筛选的结果。图6为回流温度筛选的结果。图7为料液比筛选的结果。图8为对比梯度洗脱程序一的色谱图。图9为对比梯度洗脱程序二的色谱图。图10为对比梯度洗脱程序三的色谱图。具体实施方式实施例1本发明的方法及方法学考察1材料ProminenceUFLCXR超快速液相色谱仪(日本岛津)、BSA124S电子天平(德国赛多利斯)、UPH-I-10T型超纯水器。胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷、麦角甾苷对照品均来自成都普思生物科技股份有限公司,乙腈为色谱纯,水为超纯水,其余试剂均为分析纯。独一味32批样品采自不同产地,如表1所示,经成都医学院钟世红副教授鉴定为独一味Lamiophlomisrotata(Benth.)Kudo干燥地上部分。表1独一味药材样品的来源2方法与结果2.1色谱条件Kinetex2.6μmXB-C18色谱柱,流动相乙腈(B)-0.1%磷酸水溶液(A),梯度洗脱(0~10min,6%~14.5%B;10~18.5min,14.5%~17%B;18.5~23min,17%~18%B);流速1.0mL·1.0mL·min-1;柱温30℃;进样量2μL。胡麻属苷、山栀苷甲酯、8-O-乙酰山栀苷甲酯检测波长为238nm,绿原酸、咖啡酸连翘酯苷B、麦角甾苷检测波长为320nm,木犀草苷检测波长为350nm。上述色谱条件下,样品各色谱峰保留时间适中,样品中各组分的色谱峰分离度良好,对照品及样品图见图1和图2。2.2对照品溶液制备精密称定胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷和麦角甾苷各对照品适量,加入甲醇配成含量为0.275、0.278、0.494、0.036、0.328、0.448、0.422、0.334mg·mL的混合对照品A溶液。精密吸取混合对照品A1.0mL,置于10mL容量瓶中,加入甲醇稀释至刻度,作为混合对照品B溶液。2.3供试品溶液制备取独一味药材粉末(中粉)约0.5g,精密称定,加入70%甲醇30mL,称定质量,回流提取60min,冷却后用溶剂补足重量,摇匀,过0.22μm微孔滤膜,得供试品溶液。2.4线性关系考察分别吸取2.3项下混合对照品B溶液1μL、2μL、4μL,混合对照品A溶液4μL、8μL进样2μL。结果表明上述8个成分分别在一定质量浓度与峰面积呈良好线性关系。以进样量(μg)为横坐标,峰面积为纵坐标进行线性回归,得到各对照品的回归方程、相关系数和线性范围,结果见表2。表28种成分的标准曲线成分回归方程R2线性范围/μg胡麻属苷y=713115.6111x-8510.50.99980.0275~2.200山栀苷甲酯y=900098.7648x-3933.70.99980.0278~2.224绿原酸y=2000000x+151740.99990.0494~3.952咖啡酸y=4000000x+9295.40.99980.0036~0.2888-O-乙酰山栀苷甲酯y=712731.5947x+1020.70.99990.0328~2.624连翘酯苷By=735449.0223-320.550.99990.0448~3.584木犀草苷y=1000000x+137730.99990.0422~3.376麦角甾苷y=871201.9679-2990.10.99990.0334~2.6722.5精密度试验取同一供试品(S2)2μL,重复进样5次,计算得胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷、麦角甾苷峰面积RSD分别是0.28%,0.50%,1.21%,0.50%,0.53%,0.21%,1.42%,0.16%。表明仪器精密度良好。2.6重复性试验取同一份样品(S2)粉末6份,分别按2.2项下方法制备供试品溶液,按2.1项下色谱条件测定。结果胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷、麦角甾苷峰面积RSD分别是0.49%,0.33%,1.59%,0.73%,0.64%,0.58%,0.98%,0.75%。表明方法的重复性良好。2.7稳定性试验精密吸取同一供试品溶液(S2),分别于0,1,2,4,6,12,24h时进样测定,记录峰面积。结果胡麻属苷、山栀苷甲酯、绿原酸、咖啡酸、8-O-乙酰山栀苷甲酯、连翘酯苷B、木犀草苷、麦角甾苷峰面积RSD分别是0.69%,0.66%,1.41%,1.16%,0.81%,0.55%,1.05%,0.46%。表明独一味药材供试品溶液在24h内稳定性良好。2.8加样回收率试验取同一供试品6份,每份约0.25g,精密称定,分别精密加入70%甲醇溶液配制的各对照品溶液适量,按2.3项下方法制备供试溶液并依法测定,计算加样回收率,结果见表3。表3独一味药材中8种成分加样回收率实验结果2.9样品的测定取独一味药材样品,按2.3项下方法分别制备供试品溶液,平行两份,吸取供试品溶液2μL,按2.1项下色谱条件测定。计算样品含量,结果见表4。表4独一味药材中8种成分的含量(n=2)(mg·g-1)实施例2本发明方法的工艺筛选(1)检测波长筛选对S2提取物进行全波长扫描,结果显示胡麻属苷、山栀苷甲酯、8-O-乙酰山栀苷甲酯在238nm处有最大吸收,咖啡酸323nm有最大吸收,绿原酸326nm有最大吸收,木犀草苷345nm有最大吸收,连翘酯苷B、麦角甾苷在330nm处有最大吸收,综合8种成分的吸收光谱,最终确定胡麻属苷、山栀苷甲酯、8-O-乙酰山栀苷甲酯选用238nm为检测波长,咖啡酸、绿原酸、连翘酯苷B、麦角甾苷选用320nm作为检测波长,木犀草苷选用350nm为检测波长。(2)提取方法筛选1.提取方法考察取独一味药材粉末两份(中粉)约0.5g,精密称定,加入70%甲醇30mL,称定质量,70℃,分别进行回流和超声提取60min,冷却后用溶剂补足重量,摇匀,过0.22μm微孔滤膜,得供试品溶液,按实施例1中2.1项下色谱条件测定,比较8个成分的平均峰面积,结果如图3所示。结果显示,回流提取效果较佳。2.提取溶剂浓度考察取独一味药材粉末(中粉)约0.5g,精密称定,各50%甲醇、60%甲醇、70%甲醇、80%甲醇30mL,称定质量,70℃,回流提取60min,冷却后用溶剂补足重量,摇匀,过0.22μm微孔滤膜,得供试品溶液,按实施例1中2.1项下色谱条件测定。比较8和成分的平均峰面积,结果如图4所示。结果显示,70%甲醇提取效果较佳。3.提取时间考察取独一味药材粉末(中粉)约0.5g,精密称定,加入70%甲醇30mL,称定质量,70℃,分别回流提取30min、60min、90min、120min,冷却后用溶剂补足重量,摇匀,过0.22μm微孔滤膜,得供试品溶液,按实施例1中2.1项下色谱条件测定。比较8个成分的平均峰面积,结果如图5所示。结果显示,回流提取60min效果较佳。4.提取温度考察取独一味药材粉末(中粉)约0.5g,精密称定,加入70%甲醇30mL,称定质量,分别在70℃、75℃、80℃、85℃,回流提取60min,冷却后用溶剂补足重量,摇匀,过0.22μm微孔滤膜,得供试品溶液,按实施例1中2.1项下色谱条件测定。比较8个成分的平均峰面积,结果如图6所示。结果显示,提取温度为80℃时效果较佳。5.料液比考察取独一味药材粉末(中粉)约0.5g,精密称定,分别加入70%甲醇30mL、40mL、50mL,称定质量,80℃回流提取60min,冷却后用溶剂补足重量,摇匀,过0.22μm微孔滤膜,得供试品溶液,按实施例1中2.1项下色谱条件测定。比较8个成分的平均峰面积,结果如图7所示。结果显示,料液比为1g:60mL时效果较佳。根据以上考察结果,最终确定以70%甲醇30mL为溶剂,提取温度80℃,回流提取60min此方法作为供试品制备的方法。(3)梯度洗脱程序筛选发明人对多个梯度程序进行了筛选,实验结果证明,只有在本发明洗脱工艺下才能有效分离检测出本发明要分离检测的8个成分,而采用其他的梯度洗脱工艺,比如下面的三种梯度洗脱工艺则无法有效检测:对比梯度洗脱程序一:0~8min,7%~12%B;8~19.5min,12%~17%B;19.5~23min,17%~18%B;结果如图8所示。对比梯度洗脱程序二:0~10min,6%~15%B;10~18.5min,15%~17%B;18.5~23min,17%~18%B;结果如图9所示。对比梯度洗脱程序三:0~6min,6%~8%B;6~18min,8%~18%B;18~23min,18%~18%B;结果如图10所示。综上所述,本发明的方法,通过对提取方法和色谱条件的筛选,成功建立了同时测定独一味药材中8种成分的UPLC方法,该方法准确可靠、简便快速。本发明的检测方法,为全面监控独一味药材质量提供了有效的保障。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1