一种丙炔醇醚丙烷磺酸钠含量的分析方法与流程
本发明涉及含量测量的
技术领域:
,具体而言,涉及一种丙炔醇醚丙烷磺酸钠含量的分析方法。
背景技术:
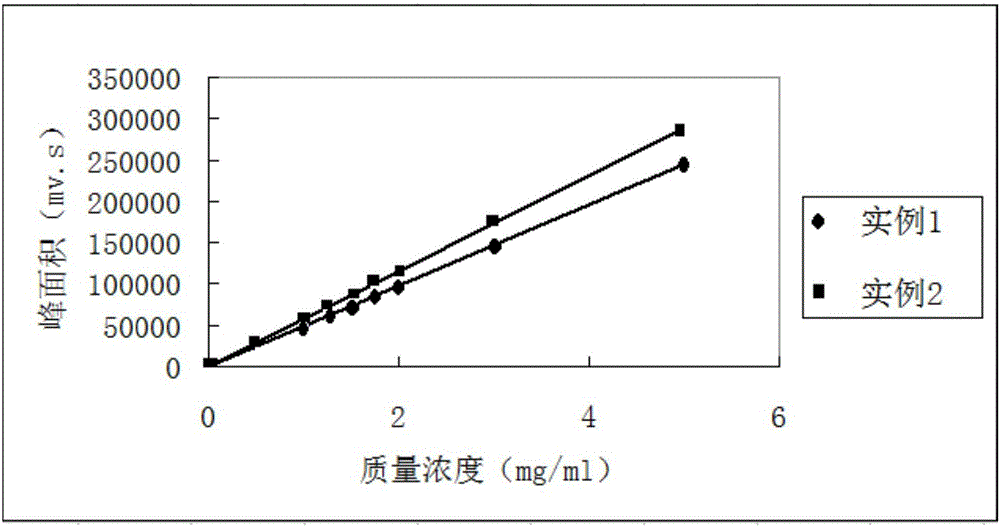
:丙炔醇醚丙烷磺酸钠(POPS),主要应用于电镀镀镍。作为镀镍中间体,属于镀镍添加剂中的主要添加成份,能起到增强低区的光亮和填平性能。在整个镀镍光亮剂市场上,此产品需求量较大,应用效果优良。在镀镍添加剂配方中,POPS的加入量涉及到配方的应用效果,光亮性、填平性、稳定性,所以POPS含量对产品质量控制是一个重要的指标,准确测定其含量很有必要。现有技术中,针对POPS含量的测定没有相关技术公开。技术实现要素:有鉴于此,本发明在于提供一种丙炔醇醚丙烷磺酸钠含量的分析方法,该分析方法准确度高,该分析方法精密度高。一种丙炔醇醚丙烷磺酸钠含量的分析方法,使用带紫外检测器的高效液相色谱采用标准曲线法获得丙炔醇醚丙烷磺酸钠含量。进一步地,所述高效液相色谱的流动相包含体积比为10~40:90~60的乙腈和缓冲溶液。进一步地,所述缓冲溶液包含1~20ml/L硝酸、1~10ml/L三乙胺和1~6g/L金属盐。进一步地,所述高效液相色谱的色谱柱的流速为0.3~1.2mL/min。进一步地,所述高效液相色谱的色谱柱的温度为10~60℃。进一步地,所述紫外检测器的检测波长为190~330nm。进一步地,使用所述高效液相色谱的标准物为百分比含量不低于48.4wt%的丙炔醇醚丙烷磺酸钠。进一步地,所述金属盐为氯化铜、醋酸汞、硝酸银和高氯酸银中的一种或至少两种。进一步地,所述高效液相色谱的流动相在使用前超声波脱气。进一步地,所述流动相在进样后在20~70℃温度下超声处理5~60min。本发明采用HPLC-UV分析法来分析丙炔醇醚丙烷磺酸钠含量。本方法具有精密度好、准确度高等优点,适用于POPS的生产过程检测和产品质量检测。附图说明图1为实施例1和实施例2的标准曲线。具体实施方式为了便于理解本发明,下面合实施例来进一步说明本发明的技术方案。如本文所用,术语:“质量份”指表示多个组分的质量比例关系的基本计量单位,1份可表示任意的单位质量,如可以表示为1g,也可表示2.689g等。假如我们说A组分的质量份为a份,B组分的质量份为b份,则表示A组分的质量和B组分的质量之比a:b。或者地,表示A组分的质量为aK,B组分的质量为bK(K为任意数,表示倍数因子)。不可误解的是,与质量分数不同的是,所有组分的质量份之和并不受限于100份之限制。“一个”、“一种”和“所述”可交换使用并指一个或多个。“和/或”用于表示所说明的情况的一者或两者均可能发生,例如,A和/或B:包括(A和B)和(A或B);另外,本文中由端点表述的范围包括该范围内所包含的所有数值(例如,1至10包括1.4、1.9、2.33、5.75、9.98等)。另外,本文中“至少一个”的表述包括一个及以上的所有数目(例如,至少2个、至少4个、至少6个、至少8个、至少10个、至少25个、至少50个、至少100个等)。作为本发明的分析方法,使用带紫外检测器的高效液相色谱采用标准曲线法获得丙炔醇醚丙烷磺酸钠含量。上述高效液相色谱(HPLC)通常包括储液器、输液泵、混合器、进样装置、分离色谱柱、检测装置五构件。上述带有紫外检测器的高效液相色谱是指检测装置的种类为紫外检测器(UV检测器或简称为UVD,以及应用紫外检测原理的二极管阵列检测器或简称为DAD)的HPLC。紫外检测器基于溶质分子吸收紫外光的原理设计的检测器,其工作原理是Lambert-Beer定律,即当一束单色光透过流动池时,若流动相不吸收光,则吸收度A与吸光组分的浓度C和流动池的光径长度L成正比相比于其它类型的检测装置,UV检测器具有灵敏度较高,线性范围宽,噪声低,波长可选,对流动相的流速和温度变化不敏感,适用于梯度洗脱,对强吸收物质检测极限可达10-9g/ml以下,检测后不破坏样品。上述标准曲线法为本领域技术人员所熟知的技术。标准曲线法标准曲线法也称为外标法或直接比较法,首先用欲测组分的标准样品绘制标准工作曲线。具体作法是:用标准样品配制成不同浓度的标准系列,在与欲测组分相同的色谱条件下,等体积准确量进样,测量各峰的峰面积或峰高,用峰面积或峰高对样品浓度绘制标准工作曲线,此标准工作曲线应是通过原点的直线。若标准工作曲线不通过原点,说明测定方法存在系统误差。标准工作曲线的斜率即为绝对校正因子。在测定样品中的组分含量时,要用与绘制标准工作曲线完全相同的色谱条件作出色谱图,测量色谱峰的峰面积或峰高,然后根据峰面积和峰高在标准工作曲线上直接查出进入色谱柱中样品组分的浓度,也可通过式(2-3-19)计算这一浓度。pi/%=fiAi(hi)(2-3-19)式中Ai(hi)——i组分峰的峰面积(峰高);fi——i组分标准工作曲线的斜率。知道进入色谱柱中样品组分的浓度后,就可根据样品处理条件及进样量来计算原样品中该组分的含量了。当欲测组分含量变化不大,并已知这一组分的大概含量时,也可以不必绘制标准工作曲线,而用单点校正法,即直接比较法定量。先配制一个和待测组分含量相近的已知浓度的标准溶液,在相同的色谱条件下,分别将待测样品溶液和标准样品溶液等体积进样,作出色谱图,测量待测组分和标准样品的峰面积或峰高,然后由式(2-3-20)直接计算样品溶液中待测组分的含量:式中ws——标准样品溶液质量分数,%;wi——样品溶液中待测组分质量分数,%;As(hs)——标准样品的峰面积(峰高);Ai(hi)——样品中i组分的峰面积(峰高)。单点校正法实际上是利用原点作为标准工作曲线上的另一个点。因此,当方法存在系统误差时(即标准工作曲线不通过原点),单点校正法的误差较大。标准曲线法的优点是:绘制好标准工作曲线后测定工作就很简单了,计算时可直接从标准工作曲线上读出含量,这对大量样品分析十分合适。特别是标准工作曲线绘制后可以使用一段时间,在此段时间内可经常用一个标准样品对标准工作曲线进行单点校正,以确定该标准工作曲线是否还可使用。上述高效液相色谱的色谱柱的型号可为UltimateXB-C18,5u,250mm×4.6mm。此处,UltimateXB-C18具有高表面覆盖率和完全封尾的特点,提高了我们色谱柱的稳定性,适合pH值1.5~10.0很宽范围内的色谱分析。可适用于中性、极性、酸性和碱性以及螯合化合物的分析具有最优的分离效率。250mm×4.6mm表示柱高为250mm,直径为4.6mm。上述高效液相色谱的流动相包含体积比为10~40:90~60的乙腈和缓冲溶液。例如乙腈和缓冲溶液的体积比10:90、12:88、15:85、20:80、30:70、35:65、38:62、40:60等。上述缓冲溶液较好地包含1~20ml/L硝酸、1~10ml/L三乙胺和1~6g/L金属盐。这里“ml/L”是指1L缓冲溶液中所含有的体积,“g/L”是1L缓冲溶液中所含有的质量。硝酸的含量可以为1ml/L、5ml/L、10ml/L、15ml/L或1ml/L等;三乙胺的含量可以为1ml/L、2ml/L、5ml/L、8ml/L、9ml/L或10ml/L等;金属盐的含量可以为1g/L、1.2g/L、1.5g/L、2g/L、3g/L、3.5g/L、4g/L、5g/L、5.5g/L或6g/L等。这里,金属盐可列举出氯化铜、醋酸汞、硝酸银和高氯酸银中的一种或至少两种。在金属盐为硝酸银的情况下,硝酸银对丙炔醇醚丙烷磺酸钠中所含有的炔键有选择性反应,即RCCH+AgNO3=RCCAg﹒xAgNO3+HNO3,在硝酸溶液中,乙炔取代物与AgNO3作用生成的产物对260-279nm范围内的单色光有最大吸收。经紫外分光光度计扫描,在未加硝酸银时,以蒸馏水做空白,在190~200nm间有最大吸收峰,且最大吸收峰随浓度增加向长波方向移动。其移动原因为,随浓度增加分子间作用显著增强,不再适用朗波-比尔定律。加入硝酸银后,以硝酸银做空白,产品在262nm处有最大吸收峰。结合Model500UV/VIS检测器,认为选用的190~200nm接近甲醇(210nm)的透过波长下限,影响检测的灵敏度。同时,低于200nm,背景噪声增大,灵敏度也会降低。所以选用产品与硝酸银反应后,最大吸收峰对应的262nm做检测波长。高效液相色谱的色谱柱的流速以0.3~1.2mL/min为宜,例如0.3mL/min、0.35mL/min、0.4mL/min、0.5mL/min、0.6mL/min、0.8mL/min、0.95mL/min、1.1mL/min、1.15mL/min或1.2mL/min等,进一步优选为0.8~1.1mL/min。高效液相色谱的色谱柱的温度较佳地为10~60℃,例如10℃、11℃、12℃、15℃、20℃、30℃、35℃、40℃、50℃、55℃或60℃等。此处色谱柱的温度即样品通过色谱柱的温度。上述紫外检测器的检测波长优选为190~330nm,例如190nm、195nm、220nm、240nm、260nm、200nm、250nm、280nm、300nm或330nm等。使用所述高效液相色谱的标准物为百分比含量不低于48.4wt%的丙炔醇醚丙烷磺酸钠。标准物作为标准曲线的标准样品的对比。高效液相色谱的流动相在使用前超声波脱气,以避免流动相中的气泡会增加基线的噪音,造成灵敏度下降,甚至无法分析。同时,还可避免溶解的氧气还所导致的某些组份被氧化,柱中固定相发生降解而改变柱的分离性能。流动相在进氧后在20~70℃温度下超声处理5~60min。例如超声处理的温度可以为20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃等;于此温度下,超声处理的时间可以为5min、15min、25min、35min、45min、55min、60min等。本发明未述及之处适用于现有技术。下面给出本发明的具体实施例,但具体实施例仅是为了进一步详细叙述本说明,并不限制本发明申请的权利要求保护范围。一下实施例所涉及的试剂和设备来源如下:实验仪器:MODEL500高效液相色谱仪(美国SSI)LabAllianceHPLCWorkstation色谱工作站超声清洗器DS-3120,天津东康科技有限公司AR2140电子天平(1/10000天平),美国奥豪斯Ultimate保护柱UltimateXB-C18色谱分离柱实施例1色谱条件:色谱柱:UltimateXB-C18,5u,250mm×4.6mm;流动相组成体积比:乙腈︰缓冲溶液=35︰65;缓冲溶液:10ml/L硝酸+5ml/L三乙胺+3g硝酸银流速:0.8mL/min;;柱温:30℃;检测器:紫外检测器;检测波长:240nm;药品:POPS:工业级,湖北吉和昌化工科技有限公司POPS:参照品,进口POPS,含量48.4%甲醇:色谱纯,国药集团化学试剂有限公司;其他试剂均为分析纯。本实施例的测定步骤如下:步骤一、缓冲溶液及流动相配制分别精确称量3g硝酸银,移取硝酸10ml,三乙胺5ml于100ml烧杯中溶解,待溶解完全后转移至1L容量瓶中,,定容后,用0.45um水系滤膜过滤待用。取0.45um有机滤膜过滤后乙腈与缓冲溶液按35︰65配制成流动相,混合均匀后,用超声清洗器加热至60℃脱气半小时冷却待用步骤二、丙炔醇醚丙烷磺酸钠标准溶液精确称取500.0mg进口POPS于50ml容量瓶中,用流动相溶解定容,该溶液浓度为10.0mg/mL。使用前用0.2um的有机滤膜过滤,清液待用。步骤三、POPS样品溶液的配制精确称取50.0mgPOPS(工业级)于50ml容量瓶中,用流动相溶解定容,该溶液浓度为1.0mg/mL。使用前用0.2um的有机滤膜过滤,清液待用。步骤四、标准曲线及线性关系精密吸取步骤2中POPS标准溶液,置于容量瓶中,流动相定容,得质量溶度为0.01mg/mL、0.1mg/mL、0.5mg/mL、1.0mg/mL、1.25mg/mL、1.5mg/mL、1.75mg/mL、2.0mg/mL、3.0mg/mL、5.0mg/mL的标准溶液,按上述色谱条件测定,每份对照液测定5次,取峰面积平均值,以质量分数为横坐标,峰面积为纵坐标,得标准曲线为y=4.92e+004x,线性范围为0.01mg/mL~5.0mg/mL,R2=1(如图1所示)步骤五、HPLC-UV法测定结果将步骤三中配制的POPS溶液,经HPLC-UV法测定,通过标准曲线计算得出:POPS的质量溶度为0.9957mg/mL,即粉末POPS质量分数为48.19%。按照以下方法对精密度及回收率试验:取质量溶度为1.0ml/mLPOPS标准溶液,重复进样7次,所得平均值为0.9625ml/mL,精密度RSD1.80%。精确量取5份质量浓度为0.5mg/mL的POPS溶液2ml,每份各加入2ml体积的0mg/mL、0.1mg/mL、0.2mg/mL、0.5mg/mL、0.7mg/mL的POPS标准溶液分别测定,取3次测定平均值计算回收率。回收率为97.1%~99.3%。上述测试结果结果见表1。表1加标回收率计算加标前量ug添加量ug加标后量ug回收量ug回收率%0.48190.4840.96590.950498.40.48190.9681.44991.416697.70.48192.422.90192.881699.30.48193.3883.86993.757797.1表中:加标前量=空白加标溶液质量溶度的测定值×进样量添加量=标准溶液质量浓度×进样量÷2加标后量=加标前量+添加量回收量=加标溶液质量浓度的测定值×进样量回收率=回收量÷加标后量×100%实施例2色谱条件:色谱柱:UltimateXB-C18,5u,250mm×4.6mm;流动相组成体积比:乙腈︰缓冲溶液=20︰80;缓冲溶液:10ml/L硝酸+5ml/L三乙胺+3g硝酸银流速:1.1mL/min;;柱温:40℃;检测器:紫外检测器;检测波长:260nm;其余实施方式均与实施例1相同。标准曲线及线性关系精密吸取步骤2中POPS标准溶液,置于容量瓶中,流动相定容,得质量溶度为0.01mg/mL、0.1mg/mL、0.5mg/mL、1.0mg/mL、1.25mg/mL、1.5mg/mL、1.75mg/mL、2.0mg/mL、3.0mg/mL、5.0mg/mL的标准溶液,按上述色谱条件测定,每份对照液测定5次,取峰面积平均值,以质量分数为横坐标,峰面积为纵坐标,得标准曲线为Y=5.796e+004X,线性范围为0.01mg/mL~5.0mg/mL,R2=1(如图1所示)。由于液相色谱的流动相比例、流速、柱温、检测波长等操作条件不同,所得标准曲线也有不同。HPLC-UV法测定结果将步骤3中配制的POPS溶液,经HPLC-UV法测定,通过标准曲线计算得出:POPS的质量溶度为0.9934mg/mL,即粉末POPS质量分数为48.08%。按照以下方法对本例的精密度及回收率试验:取质量溶度为1.0ml/mLPOPS标准溶液,重复进样7次,所得平均值为0.9500ml/mL,精密度RSD2.65%。精确量取5份质量浓度为0.5mg/mL的POPS溶液2ml,每份各加入2ml体积的0mg/mL、0.1mg/mL、0.2mg/mL、0.5mg/mL、0.7mg/mL的POPS标准溶液分别测定,取3次测定平均值计算回收率。回收率为96.7%~98.9%,结果见表2。表2加标回收率计算加标前量ug添加量ug加标后量ug回收量ug回收率%0.48190.4840.96590.945697.90.48190.9681.44991.434098.90.48192.422.90192.826597.40.48193.3883.86993.742296.7表中:加标前量=空白加标溶液质量溶度的测定值×进样量添加量=标准溶液质量浓度×进样量÷2加标后量=加标前量+添加量回收量=加标溶液质量浓度的测定值×进样量回收率=回收量÷加标后量×100%由于本发明中所涉及的各工艺参数的数值范围在上述实施例中不可能全部体现,但本领域的技术人员完全可以想象到只要落入上述该数值范围内的任何数值均可实施本发明,当然也包括若干项数值范围内具体值的任意组合。此处,出于篇幅的考虑,省略了给出某一项或多项数值范围内具体值的实施例,此不应当视为本发明的技术方案的公开不充分。申请人声明,本发明通过上述实施例来说明本发明的详细工艺设备和工艺流程,但本发明并不局限于上述详细工艺设备和工艺流程,即不意味着本发明必须依赖上述详细工艺设备和工艺流程才能实施。所属
技术领域:
的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式选择等,落在本发明的保护范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1