本发明涉及基于“一测多评”法的花椒麻味物质含量检测。
背景技术:
花椒的麻味物质主要集中在花椒果皮的油腺细胞,研究表明,花椒中的不饱和脂肪酸酰胺类成分为花椒中的麻味物质。目前,已有学者从花椒中鉴定出了25种麻味物质,包括α-山椒素,β-山椒素,γ-山椒素,羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素等。其中,羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素是花椒中最主要的麻味物质。
花椒的麻味是花椒品质评价的主要的表现形式之一,目前仍没有市售的花椒麻味物质对照品,并且现行的花椒国家标准和地方标准没有对花椒的麻味进行客观定量评价。
因此,有必要对花椒中麻味成分进行分离,通过同时测定多个麻味物质指标性成分,实现对花椒中的麻味物质含量检测。
技术实现要素:
为解决上述问题,本发明提供基于“一测多评”法的花椒麻味物质含量检测,包括以下步骤:
(1)羟基-β-山椒素标准曲线的建立:
a、对照品溶液的制备:
取羟基-β-山椒素对照品,加甲醇配制成对照品溶液;
b、对照品溶液的测定:
配制系列浓度的对照品溶液,分别注入高效液相色谱仪,测定各色谱峰峰面积,得到羟基-β-山椒素的标准曲线;
色谱条件如下:
检测波长:270±5nm;
色谱柱:C18色谱柱;
流动相:乙腈:水=40%:60%;
(2)待测样品中羟基-β-山椒素的含量测定:
c、供试品溶液的制备:
取待测样品,加甲醇提取,过滤得供试品溶液;
d、供试品溶液的测定:
取供试品溶液,注入高效液相色谱仪,以步骤b相同的色谱条件检测,根据步骤(1)的标准曲线得到待测样品中羟基-β-山椒素的含量;
(3)使用相对校正因子计算待测样品中羟基-α-山椒素和羟基-γ-山椒素含量。
进一步地,所述C18色谱柱为Phenomenex C18色谱柱、Waters Bridge C18或Agilent Eclipse Plus C18色谱柱。
进一步地,所述色谱柱的规格为250mm×4.6mm,5μm。
进一步地,所述高效液相色谱仪为Agilent 1260、岛津LC-2010A或Waters e2695。
进一步地,步骤c中,所述提取是超声提取。
进一步地,所述超声提取的时间为30分钟。
进一步地,步骤c中,所述甲醇与待测样品的体积质量比为100mL:1g。
进一步地,所述色谱条件的柱温为20~30℃,优选30℃。
进一步地,所述色谱条件的流速为0.6~1.0mL·min-1,优选1.0mL·min-1。
进一步地,所述步骤(3)的相对校正因子是通过下述步骤得到的:
取羟基-α-山椒素、羟基-γ-山椒素和羟基-β-山椒素对照品,加甲醇配制成混合对照品溶液;
取混合对照品溶液,注入高效液相色谱仪,以步骤b相同的色谱条件检测,得到羟基-α-山椒素、羟基-γ-山椒素和羟基-β-山椒素的标准曲线;
以羟基-β-山椒素为内参物,根据相对响应因子f计算公式:计算羟基-α-山椒素、羟基-γ-山椒素与羟基-β-山椒素之间的相对应因子,取其均值;
其中,相对响应因子f计算公式如下:
式中A为色谱峰面积、W为成分的质量浓度,下标n代表待测成分,下标s代表内参物。
本发明还提供了本发明的含量检测用于检测花椒、花椒油、藤椒、藤椒油及其产品中麻味物质的含量检测。
经试验证明,本发明色谱条件可以有效将羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素分离,从而准确测定它们的含量。并且,本发明的“一测多评”法与外标法相互验证,无明显差异,结果可靠。
另外,发明人分离制备得到的羟基-β-山椒素对照品已被中检所授权,可以作为花椒的对照品使用。从而本发明方法可以克服在缺乏羟基-α-山椒素及羟基-γ-山椒素对照品的情况下通过相对校正因子f及色谱峰定位计算其含量,实现花椒中主要麻味物质羟基-α-山椒素成分及羟基-γ-山椒素质量控制,为花椒的质量控制提供了新的方法。因此,本发明的方法可以用于花椒、藤椒及其产品的检测。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
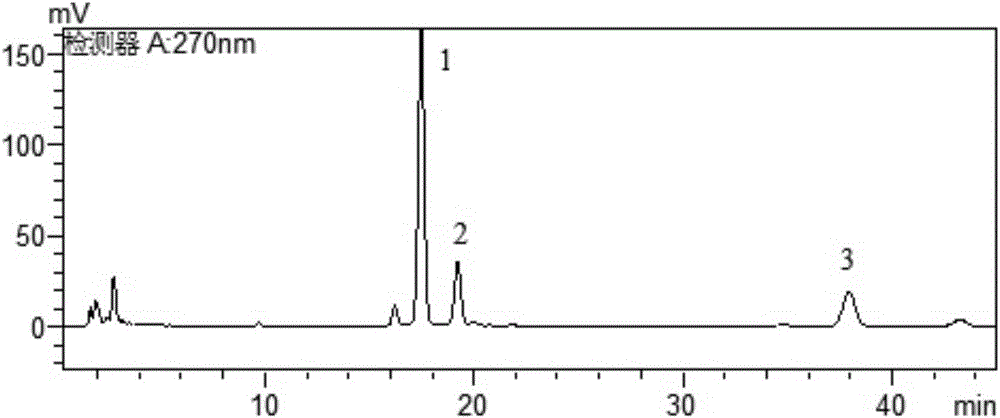
图1为混合对照品溶液HPLC色谱图(1—羟基-α-山椒素;2—羟基-β-山椒素;3—羟基-γ-山椒素)
图2为供试品溶液HPLC色谱图(1—羟基-α-山椒素;2—羟基-β-山椒素;3—羟基-γ-山椒素)
具体实施方式
实施例1
1仪器
Agilent 1260高效液相色谱仪,G1311C四元泵,G1329B自动进样器,G1316A柱温箱,G1362A二极管阵列检测器(美国Agilent公司);Waters e2695高效液相色谱仪,四元泵,2998PDA检测器;Shimadzu岛津LC-2010A,SIL-20A自动进样器,SPD-20A检测器,CTO-20A柱温箱,LC solution色谱工作站软件(日本岛津公司);BP211D电子分析天平(德国Sartouris股份有限公司)。
2材料和试剂
花椒:四川汉源花椒局提供。
花椒麻味物质对照品:羟基-α-山椒素(自制,纯度98.35%);羟基-β-山椒素(自制,已申请成国家对照品,纯度98.27%);羟基-γ-山椒素(自制,纯度98.54%)。
甲醇:色谱级,美国sigma公司。乙腈:色谱级,美国sigma公司。
3 HPLC色谱条件
色谱柱Phenomenex C18柱(250mm×4.6mm,4μm);以乙腈(A):水(B)=40%:60%为流动相,等度洗脱45min,柱流量1.0min/mL,检测波长270nm,柱温30℃,进样量10μL。
4一测多评法的建立
4.1相对校正因子的确定
4.1.1混合对照品溶液的制备
分别精密称定羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素对照品适量置于100mL容量瓶中,加甲醇定容至刻度,配成质量浓度分别为1.70mg/mL,0.475mg/mL,0.189mg/mL的对照品溶液,作为对照品储备溶液。精密吸取上述对照品储备液各1mL,置10mL容量瓶中,加甲醇定容至刻度,摇匀,配成对照品中间液。然后,分别精密移取上述对照品中间液各2mL,置10mL容量瓶中,加甲醇定容至刻度,摇匀,配成混合对照品溶液。将上述混合对照品溶液保存于4℃冰箱中备用,备用。
4.1.2供试品溶液制备
精密称取花椒粉(过四号筛)约0.5g精确至(0.0001g),置具塞锥形瓶中,加入50mL甲醇,超声振荡30min过滤后即得。分别精密吸取上述对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,结果见图1、图2。
4.1.3线性、定量限与检测限
分别精密吸取4.1.1项下混合对照品溶液2、5、10、15、20、25、30μL进样分析,以峰面积为纵坐标,进样浓度为横坐标,绘制标准曲线;以最低浓度的对照品溶液逐级稀释,分别以信噪比为3和10的浓度作为检测限和定量限,见表1。
表1线性关系及定量限、检测限
4.1.4相对校正因子的计算
在“2”项下确定的HPLC色谱分析条件下,精密吸取“4.1.1”项下的混合对照品溶液,分别进样2、5、10、15、20、25、30μL。以羟基-β-山椒素为内参物,根据相对响应因子(f)计算公式(1),计算羟基-α-山椒素、羟基-γ-山椒素与羟基-β-山椒素之间的相对应因子,取其均值。
式中A,W,分别为色谱峰面积、成分的质量浓度,下标n,s分别代表待测成分及内参物。结果见表2,该条件下的计算结果相对校正因子值为f羟基-α-山椒素/羟基-β-山椒素=1.0238,RSD为0.87%;f羟基-γ-山椒素/羟基-β-山椒素=0.7986,RSD为0.54%表明不同进样体积下的相对校正因子较为稳定。
表2不同进样体积下的相对校正因子
4.1.5精密度试验
精密移取“4.1.1”项下混合对照品溶液,连续进样6次,每次10μL,分别记录各成分峰面积并计算其RSD,结果羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素的RSD值分别为0.16%,0.11%,0.32%,表明仪器精密度良好。
4.1.6重复性试验
精密称取同一批花椒粉末约0.5g,按3.1.2项下操作,平行制备6份供试品溶液,测得羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素的含量RSD分别为0.02%,0.42%,0.29。表明样品重复性良好。
4.1.7稳定性试验
精密吸取同一混合对照品溶液10μL,分别于0,2,4,6,8,12,24,36h进样,分别测定羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素的峰面积,其峰面积的RSD分别为0.25%,0.27%,0.47。表明该混合对照品溶液在36h内稳定性良好。
4.1.8加样回收试验
取同一批已知含量的花椒粉末0.25g,精密称定,平行6份,分别精密加入羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素对照品储备溶液,按4.1.2项下制备供试品溶液测定,计算羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素的加样回收率,结果见表3。
表3花椒中羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素的加样回收率试验结果
4.2一测多评法系统耐用性和系统适用性
4.2.1相对校正因子的耐用性和系统适用性
本实验分别选用Agilent 1260、岛津LC-2010A及Agilent 1200高效液相色谱系统与Agilent Eclipse Plus C18(4.6mm×250mm,5μm)、Waters Bridge C18(4.6mm×250mm,5μm)及Phenomenex C18(4.6mm×250mm,5μm)3种色谱柱,考察色谱系统对相对校正因子的影响。结果显示不同色谱系统对其影响较小(RSD<5.0%),重现性良好,见表4。
表4不同色谱系统对相对校正因子的影响
4.2.2待测组分色谱峰的定位
4.2.2.1不同仪器和色谱柱对相对保留时间
根据羟基-β-山椒素的保留时间,利用羟基-α-山椒素相对于羟基-β-山椒素的相对保留时间r,见公式(2),即可对峰进行定位。
r=rx/rs (2)
其中rx,rs分别表示待测物和内参物的保留时间。
实验分别考察了相对保留时间值在不同品牌色谱仪、不同品牌色谱柱的重现性,结果见表5。结果表明,不同色谱柱对各成分的保留时间变化较明显,但各成分间相对保留值变化不大,RSD<5.0%,同型号色谱柱在不同仪器上各成分的保留时间变化不大。因此,本实验选用相对保留值作为目标色谱峰的定位指标较为合适。
表5一测多评法待测成分色谱峰的定位
4.2.2.2不同流速和柱温对相对保留时间
在相同条件下,采用Agilent 1260色谱系统和Waters Bridge C18色谱柱分别考察不同流速(0.6、0.8、1.0mL·min-1)和不同柱温(20,25,30℃)对羟基-α-山椒素、羟基-β-山椒素的相对保留时间的影响,结果显示不同流速、柱温对相对保留时间的影响较小(RSD<3.0%)。
4.3“一测多评”法外标法比较
首先采用外标法对花椒中羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素进行测定,再建立一测多评法进行计算,并将两种方法计算的结果进行比较,以验证一测多评法对用于测定花椒中麻味物质评价的准确性。结果表明,两种方法测得的花椒麻味物质含量无显著性差异,提示建立的方法具有较好的可信度。结果见表6。
表6外标法和一测多评法测定花椒中羟基-α-山椒素含量的比较
5讨论
5.1检测波长
本实验选用高效液相DAD检测器,在190~400nm下对制备的羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素对照品进行了全波长扫描。结果显示羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素对照品最大吸收分别在272nm,267nm,270nm,处。考虑到羟基-γ-山椒素含量最低,故选择其最大吸收270nm作为检测波长。
5.2内参物的选择
本实验选用羟基-β-山椒素为内参物,因其对照品制备易得,理化性质较为稳定,可以长期保存使用,已被中检所复核批准作为花椒对照品使用。
5.3色谱条件的选择
羟基-α-山椒素、羟基-β-山椒素为顺反式同分异构体,性质相似、较难分离。笔者尝试用甲醇-水、乙腈-水、甲醇-水-甲酸、乙腈-水-甲酸等流动相系统在等度洗脱的条件下对羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素的分离进行考察,结果显示采用乙腈-水系统(40:60)进行等度洗脱,分析时间较短,两个成分均能达到理想分离,且峰形良好。故最终采用乙腈-水系统(40:60)作为流动相进行等度洗脱。
5.4结果分析
本发明测得了12批花椒样品中麻味物质羟基-α-山椒素,羟基-β-山椒素,羟基-γ-山椒素的含量。本发明采用的“一测多评”法与外标法相互验证,无明显差异,结果可靠。本发明方法可以克服在缺乏羟基-α-山椒素及羟基-γ-山椒素对照品的情况下通过相对校正因子f及色谱峰定位计算其含量,实现花椒中主要麻味物质羟基-α-山椒素成分及羟基-γ-山椒素质量控制。本发明建立的“一测多评”法为花椒的质量控制提供了新的方法。