IC‑ICP‑MS联用测定茶叶中砷形态的方法及应用与流程
本发明涉及一种离子色谱-电感耦合等离子体质谱联用测定茶叶中砷形态的方法及应用。
背景技术:
茶是一种常用饮料,茶叶中除含有茶多酚、咖啡碱、维生素等对人体有益的有机成分外,还含有多种微量元素。微量元素对人体是有益的,也有少数元素是有害的,必须在种植生产和加工过程中对其加以有效监测和控制。准确测定茶叶中的微量元素,对于评价茶叶质量有重要的意义。
研究发现,砷污染严重危害人体健康,但砷对人及生态系统的毒性不仅与砷总量有关,还与砷存在的化学形态密切相关。在自然界中,砷的主要形态有砷酸盐(As(Ⅴ)、亚砷酸盐As(Ⅲ)、一甲基砷(MMA)、二甲基砷(DMA)、砷甜菜碱(AsB)、砷胆碱(AsC)和砷糖(AsS)等,其中As(Ⅴ)、As(Ⅲ)为无机砷,其他均为有机砷。无机砷的毒性很高,已有研究证明无机砷为致癌物质,而有机形态砷大多无毒。由于砷的各种形态随着所处环境的不同始终处转化之中,所以单独测定总砷含量已无法准确反映砷的暴露水平及其对环境和生态的影响。因此,对砷的存在形态进行分析测定十分必要。
当前研究中,有关砷的形态研究大多集中在土壤环境、水质、海产品和少数其他食品中,其中砷化合物的提取溶剂大多采用甲醇-水混合液或稀酸溶液(车东昇等,环境化学,2014,2014,33(7): 1130-1136)。目前行业标准《NY 659-2003》仅对茶叶中的砷总量做出限定,对茶叶中各种砷形态进行全面的调查分析是目前的难点。
技术实现要素:
为了克服现有技术的不足,本发明提供了一种IC-ICP-MS联用测定茶叶中砷形态的方法及应用。
一种IC-ICP-MS联用测定茶叶中砷形态的方法,
1)待测茶叶经过干燥、粉碎、过筛,利用0.1mol·L-1硝酸水溶液提取,石墨消解仪100℃消解2h,得到待测液;
2)利用离子色谱进行分离待测液;
3)利用ICP-MS采用CCT模式测定砷形态。
所述的硝酸水溶液提取条件具体如下,称取粉碎的样品1.00 g于石墨消解仪的50 mL特氟龙消解罐中,加10 mL的0.2 mol·L-1硝酸溶液,混匀,90℃消解2h,8000 r·min-1离心15 min,取上清液过滤得到待测液。
所述的离子色谱条件:
Dionex IonPac AS7,250mm×2mm;
流动相,A为5 mol·L-1 (NH4)2CO3,B为100 mol·L-1 (NH4)2CO3;
二元梯度程序:0~2.5 min,100%A;2.5~5min,100% B;5~15min,100% A;流速0.3 mL·min-1,进样体积25 μL。
所述的 ICP-MS条件:
功率 1400 W;
冷却气流速13.0 L·min-1;
辅助气流速0.88 L·min-1;
载气流速1.12 L·min-1;
采样深度150;
四极杆偏压-6.5 v;
六极杆偏压-8.5 v;
聚焦电压9.22 v;
采集时间800 s;
碰撞气流量3.0 L·min-1。
所述的方法的应用,根据不同砷形态组成,应用于茶叶产地溯源研究。
本发明的有益效果是,该方法检测速度快、准确度高,成本低,适用于实际样品的检测,可用于茶叶中四种砷形态的准确定量和风险评估。
由于茶叶中砷形态与产地(包括气候土壤等大环境)相关,也与人工管理和茶叶加工有关,所以可以应用于茶叶产地溯源研究。
附图说明
下面结合附图和实施例对本发明进一步说明。
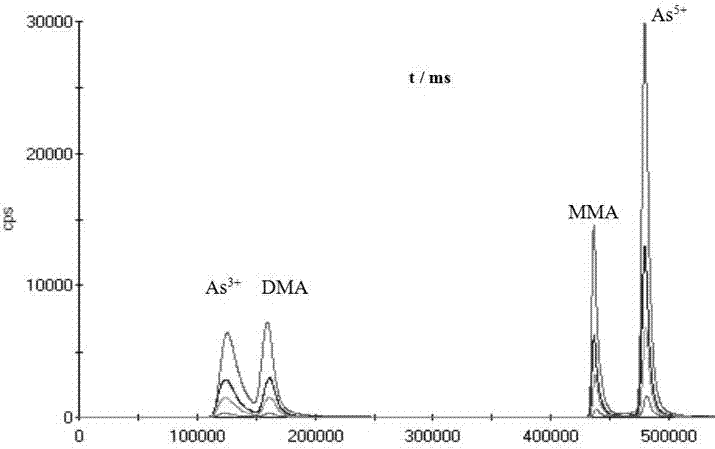
图1是20 μg•L-1的As(Ⅲ)、DMA、As(V)和MMA分离图;
图2是4个砷形态0.5-25 μg·L-1标准曲线谱图。
具体实施方式
本发明采用硝酸提取,以Dionex IonPac AS7阴离子交换柱为分离柱,在相关研究(Chungang Yuan etc. ,Food and Chemical Toxicology, 2007(45): 2381-2389)的基础上,对试验条件进行了优化,结合ICP-MS检测,成功地建立了茶叶中4种主要砷形态的检测方法,并通过对4个茶类中砷形态的分析,对茶叶中砷形态现状作了初步探索,为茶产品种植加工过程中砷变化规律的研究和茶叶品质监测提供技术支持,为研究茶产品中砷的风险评估奠定基础。
材料与方法
1.1 茶样
4个茶类共63个样品均取自市售,其中花茶10个来自新疆,砖茶20个取自湖北,绿茶18个取自浙江,红茶15个取自云南。样品称量、消解前均经过干燥、粉碎、过筛。
仪器与试剂
仪器:Thermo Fisher XSeries II型电感耦合等离子体质谱仪(美国Thermo Fisher公司);ICS-3000型离子色谱(美国Thermo Fisher公司);Milli-Q超纯水处理系统(美国 Millipore公司);DEENA II样品全自动消解前处理系统(美国Thomas Cain),IR210旋转蒸发仪(瑞士Buchi公司);Z383K型离心机(德国Hermle公司);KQ-100超声波清洗器(江苏省昆山市超声仪器有限公司);Buchi Waterbath B-480旋转蒸发仪(瑞士 Buchi 公司)。
试剂:亚砷酸钠As(Ⅲ),砷酸钠As(V),一甲基砷酸钠(MMA)和二甲基砷酸(DMA),均购于Fluka公司。碳酸铵为优级纯,阿拉丁试剂有限公司;甲醇和过氧化氢,默克公司;水为超纯水。用超纯水配制1.0 mg·L-1的混合As(Ⅲ)、DMA、MMA和As(Ⅴ)(以砷计)砷形态储备液;工作溶液使用超纯水逐级稀释配制,现用现配。流动相使用前经超声波清洗器超声脱气。
样品前处理
1.2.1 砷总量的测定
准确称量0.2 g左右(精确至0.0001 g)茶叶样品于特氟龙消解罐中,加入5mL硝酸和1 mL双氧水,微波消解,参数见表1。消解结束后,将样品转移至干净的容量瓶中,稀释定容至25mL,供ICP-MS测定。
1.2.2 不同形态砷的测定
采用三种前处理方法对茶叶中存在的砷形态进行提取。
方法一:准确称取粉碎的样品1.00 g于15 mL具塞离心管中,加10 mL的甲醇-水(体积比1:1),混匀,超声提取30 min,8000 r·min-1离心15min,取上清液过滤。旋转蒸发仪将滤液浓缩,减少甲醇的对柱子的伤害。用超纯水定容至25mL,即可用于砷的形态分析。
方法二:准确称取粉碎的样品1.00 g于50 mL特氟龙消解罐中,加10 mL的0.2 mol·L-1硝酸溶液,混匀,90℃消解2h,8000 r·min-1离心15 min,取上清液过滤。用超纯水定容至25 mL,即可用于砷的形态分析。
方法三:准确称取粉碎的样品1.00 g于50 mL特氟龙消解罐中,加入25mL沸水,100℃石墨消解仪保持微沸30 min,冷却,8000 r·min-1离心15 min,取上清液过滤。用超纯水定容至25 mL,即可用于茶汤中砷形态分析。
混合标准溶液的配制
准确称取4种砷形态的化合物,用去离子水配成含砷1.0 mg·L-1的母液,置于4℃冰箱中保存备用。工作溶液使用去离子水逐级稀释配制,现用现配。流动相使用前经超声波清洗器超声脱气。
仪器工作条件
IC条件:Dionex IonPac AS7(250mm×2mm) 阴离子交换柱;流动相:A为5 mol·L-1(NH4)2CO3,B为100 mol·L-1 (NH4)2CO3。二元梯度程序:0~2.5 min,100%A;2.5~5min,100% B;5~15min,100% A;流速0.3 mL·min-1,进样体积25 μL。
ICP-MS条件:以1.0 μg·L-1的7Li、59Co、115In、238U调谐液进行仪器条件最佳化选择,使1.0 μg·L-1的115In和238U的计数分别大于4.0×104 cps和8.0×104 cps以进行全质量范围内质量校正。氧化物比率CeO+/Ce+≤0.5%,双电荷比率Ba++/Ba+≤2%;由于35Cl和仪器所用的高纯载气(氩气)易形成75ClAr+,干扰75As的测定,因此,采用时间CCT模式,用以消除75ClAr+干扰。ICP-MS的工作参数分别见表2。
数据处理
由保留时间定性,峰面积定量。绘制标准曲线,外标法定量,计算各个砷形态的含量。以Excel和SPSS 19.0进行作图和数据处理。单因素方差分析(Analysis of variance,ANOVA)用来比较各组间的差异,P<0.05表示差异性显著。
结果与讨论
2.1 色谱条件优化
2.1.1 流动相
比较了(NH4)2CO3、硝酸:甲醇混合溶液(体积比为95:5)、硝酸、作为淋洗液对As(Ⅴ)、As(Ⅲ)、MMA、DMA的洗脱效果。结果显示,当(NH4)2CO3和硝酸甲醇混合溶液作为流动相时,As(Ⅴ)、As(Ⅲ)、MMA、DMA完全分离,且相同浓度的硝酸甲醇混合溶液比(NH4)2CO3的分析时间短,但As(Ⅲ)和DMA分离效果较差,硝酸的分离效果更差。因此,选择(NH4)2CO3为本试验流动相。
浓度及流速
分析了1,3,5,10 mmol·L-1的碳酸铵在指定流速下对As(Ⅲ) 和DMA的分离效果,结果显示:随着(NH4)2CO3浓度的增大,As(Ⅲ) 、DMA的保留时间减少,选择浓度10 mmol·L-1时两者重叠;选择1 mmol·L-1时,As(Ⅲ) 有拖尾严重;选择3 mmol·L-1和5 mmol·L-1时,分离度相差不大,但出峰时间显著差异,因此选择5 mmol·L-1 (NH4)2CO3作为As(Ⅲ) 、DMA的分离浓度。考察了20、100、200 mmol·L-1的(NH4)2CO3在指定流速下对As(V)和MMA的分离效果,结果显示:随着洗脱浓度的增加,两者出峰时间缩短,但分离度降低,因此选择浓度适中的100 mmol·L-1 (NH4)2CO3作为As(V)和MMA的洗脱浓度。
分别比较了流速为0.1、0.3、0.5 mL·min-1时的出峰情况及柱压,当流速为0.5 mL·min-1时,保留时间减少,但As(Ⅲ) 、DMA分离度降低,且柱压超过4000 psi,不利于保护柱子;当流速为0.1 mL·min-1时,分离时间延长,并且峰拖尾显著,因此选择0.3 mL·min-1为本试验最佳流速。
综合考虑选择流速为0.3 mL·min-1,(NH4)2CO3浓度为5 mmol·L-1和100 mmol·L-1作为流动相梯度分离测定As(Ⅲ)、DMA、As(V)和MMA。先用4种单标进样确定其出峰时间,再用混合标准溶液,进一步确定各物质的出峰顺序。图1为20 μg·L-1的As(Ⅲ)、DMA、As(V)和MMA分离图,4种物质得到较完全分离,其保留时间分别约为2.08、2.92、7.34、8.03 min。
样品提取方法优化
目前针对砷形态的提取溶剂主要有水、酸液、甲醇和酶等试剂。本研究比较了100℃水煮30min、甲醇-水(1:1)超声30min和0.2 mol·L-1硝酸水90℃消解2h三种提取方式对4种砷形态的提取效率。上述三种提方式对4种砷形态的提取效果见表3。
由表3可知,以0.2 mol·L-1硝酸水溶液对茶叶中砷的提取效率在78%~88%之间;水煮提取最低,均不超过50%;甲醇水溶液提取居于中间,在53%~68%之间。由于甲醇水溶液提取后,提取液须旋蒸或氮吹将甲醇尽可能去除,以避免对分析柱的损害,因此该方法相对繁琐。综合考虑选择以0.2 mol·L-1硝酸水溶液100℃消解2h作为茶叶中砷形态的提取方法。
标准工作曲线、检出限及方法准确性
在优化条件下,采用逐级稀释法配制质量浓度为0.5、1、5、10、25 μg·L-1的4种砷系列标准混合溶液,绘制标准曲线,将s/n =3时的样品浓度作为仪器检出限(LOD),s/n=10时的样品浓度作为仪器定量下限(LOQ)。结果如表3所示,4种砷形态在0.5~25 μg·L-1范围内呈良好的线性关系良好( r2 ≥0.999 ),As(Ⅲ)、As(V)、MMA和DMA 4种砷形态的检出限(LODs)分别为0.2、0.1、0.3、0.3 μg·L-1,定量下限(LOQs)分别为0.5、0.3、0.5、0.6 μg·L-1。对1 μg·L-1砷混合标准溶液进行24h内和48h内相对标准偏差(RSD)的测定,得到4种砷形态的24h内RSD(n=6)小于4.1%,48h内RSD(n=3)小于4.6%,两项指标均可满足日常检测需求。
2.5 加标回收率与方法稳定性实验
在3个加标水平下,测定IC-ICP-MS方法的回收率及稳定性,结果如表5所示。4种砷形态在1.0、5.0和10.0 μg·kg-1三个加标水平下的回收率为90.9%~125.0%。实验重复4次,其相对标准偏差小于5.7%,可用于定量分析。4个砷形态0.5-25 μg·L-1标准曲线谱图如图2所示。
2.4 茶叶中As(Ⅲ)、DMA、MMA、As(Ⅴ)的分析结果
将4个茶类共63个样品前处理后进行分离与检测,结果显示(见表6-1,6-2)。63个样品中共有7样品未检出砷,其中绿茶4个,红茶3个。绿茶、红茶、砖茶、花茶中砷总量平均值分别为43.5、42.8、303.7、573.9 μg·kg-1,可提取总量分别为38.0、38.0、285.0、509.7 μg·kg-1。绿茶中只检出As(V),红茶有3只样品检出少量As(III),其余只检出As(V),两个茶类中均未检出有机形态砷。砖茶和花茶中均检出As(V)和As(III),且含量相对较高,其中砖茶和花茶中4种砷形态As(III)、As(DMA)、As(MMA)、As(V)占对应提取总量的百分比分别为24.8%、8.9%、10.2%、56.1%和23.1%、7.7%、10.0%、59.2%。根据上述数据分析,茶叶中无机砷含量显著高于有机砷(P<0.05),并以无机形态的As(V)为主,含量从高到低依次为As(V)、As(Ⅲ )、MMA、DMA,该情况与烟草类似。
4 结论
本研究建立的离子色谱-电感耦合等离子体质谱联用分析方法,并对茶叶中砷的提取方法和流动相体系进行了优化,实现茶叶中As(Ⅴ)、As(Ⅲ)、MMA和DMA四种砷形态的同时检测。该方法检测速度快、准确度高,适用于实际样品的检测,同时对市售4个茶类中砷背景值及4种砷形态含量进行了初步分析,为进行茶叶中砷残留检测及风险评估提供了基础数据。
- 还没有人留言评论。精彩留言会获得点赞!