一种定量确定微生物中表达的目的蛋白的表达情况的方法与流程
本方法发明属于蛋白表达领域,涉及一种定量确定微生物中表达的目的蛋白的表达情况的方法。
背景技术:
在生物蛋白研发及生产过程中,都会涉及到蛋白表达情况的检测,包括可溶性目的蛋白的表达量,以及可溶性蛋白与包涵体的比例等等。而现如今大部分生物科研院所和生物公司,对蛋白表达情况的检测,主要是定性的检测,而且定性检测效果也不尽如意,比如,蛋白电泳结果不是很糊分不清,就是很淡看不见。严重影响对蛋白表达情况的本质把握,不能正确指导后续蛋白表达优化,蛋白放大生产和后续的蛋白分离纯化工作,从而造成时间,人力及资源的浪费。
技术实现要素:
有鉴于此,本发明目的是提供一种操作简单方便,所用到的仪器设备都是生物实验室常用仪器。从定量的角度上来分析蛋白表达情况,解决现有问题,为后续蛋白表达优化,蛋白放大生产及分离纯化提供可靠依据的定量确定微生物中表达的目的蛋白的表达情况的方法。
为了解决上述技术问题,本发明的技术方案是:
一种定量确定微生物中表达的目的蛋白的表达情况的方法,对于一表达了目的蛋白的微生物菌液,总体积为v总ml,其在波长为600nm的光照下的光吸收值od600为a,从v总中取出的用于检测目的蛋白表达情况的菌液体积为v检ml。那么,取出的菌液经离心去上清液后,得到菌体沉淀,往菌体沉淀中加入的裂解缓冲液体积为40v检aul。
进一步的,将所述体积为40v检aul的含菌体沉淀的裂解缓冲液混匀,并用超声破碎仪将菌体破碎后,各取30ul的破菌液于两个1.5mlep管中。其中一个ep1管中加入10ul的4*蛋白样品处理液,用于跑变性蛋白电泳的全菌液样品s全菌;另一个ep2管于高速离心机中离心,将离心后的上清液小心转入另一个新的ep3管中,并向ep3管中加入10ul的4*蛋白样品处理液,用于跑变性蛋白电泳的上清液样品s上清;已移除上清液的ep2管中所剩下的沉淀即为包涵体,往已移除上清液的ep2管中包涵体加入与上清液体积相同的裂解缓冲液30ul,并向ep2管中加入10ul的4*蛋白样品处理液,用于跑变性蛋白电泳的包涵体样品s包涵体。
进一步的,所述s全菌,s上清,s包涵体,按同样方法制得的不含目的蛋白的全菌液样品s阴性和已知蛋白浓度的纯蛋白s定量连同蛋白maker一起跑变性蛋白电泳,上样量都为v上样ul。根据蛋白电泳胶图结果,可看出目的蛋白在上清液和包涵体中的大致分布,目的蛋白是在上清液中多,还是在包涵体中更多,此即为定性。通过凝胶成像系统分析蛋白电泳胶图,可得出s全菌,s上清,s包涵体中各自目的蛋白的量与s定量中已知蛋白量的比例,从而得到上样量都为v上样ul的s全菌,s上清,s包涵体里目的蛋白的量分别为m全菌ug,m上清ug,m包涵体ug,其中m全=m上清+m包涵体。因此,上清液中目的蛋白量与包涵体中目的蛋白总量的比值为d=m上清/m包涵体,其中上清液中的目的蛋白即为可溶性目的蛋白,从而体积为v总ml的微生物菌液中可溶性目的蛋白的总量m上清=160m上清v总a/(3v上样)ug;体积为v总ml的微生物菌液中可溶性目的蛋白的浓度为d上清=160m上清a/(3v上样)ug/ml。此即为定量分析。
进一步的,所述不含目的蛋白的全菌液样品s阴性,即做为实验的阴性对照,用于判断目的蛋白是否在实验组有表达。
进一步的,所述已知蛋白浓度的纯蛋白s定量,即做为实验的定量标尺,用于定量目的蛋白的量。
进一步的,所述d值越大,上清液中的目的蛋白越多,也就是可溶性目的蛋白越多;当d>1时,上清液中的目的蛋白比包涵体中的目的蛋白多,也就是可溶性目的蛋白更多。
进一步的,所述d上清=160m上清a/(3v上样)ug/ml=60m上清a/(3v上样)mg/l,表示每l菌液中含可溶性目的蛋白的毫克数。以此来判断目的蛋白的表达是否可观。
进一步的,所述定量分析是通过凝胶成像系统来分析蛋白电泳胶图,对于无凝胶成像系统的实验室,也可通过肉眼大致得出目的蛋白与定量蛋白之间的比例,从而得出粗略的相应数据。
本发明技术效果主要体现在以下方面:本方法发明操作简单方便,所用到的仪器设备都是生物实验室常用仪器。从定量的角度上来分析蛋白表达情况,解决现有问题,为后续蛋白表达优化,蛋白放大生产及分离纯化提供可靠依据。
附图说明
图1为本发明的实施例的蛋白电泳胶图;
图2为本发明的对照组1电泳胶图;
图3为本发明的对照组2电泳胶图;
图4为本发明的对照组3电泳胶图。
具体实施方式
以下结合附图1-4,对本发明的具体实施方式作进一步详述,以使本发明技术方案更易于理解和掌握。
实施例
本方法发明操作简单方便,所用到的仪器设备都是生物实验室常用仪器。从定性和定量的角度上来分析蛋白表达情况,解决现有问题,为后续蛋白表达优化,蛋白放大生产及分离纯化提供可靠依据。
对于一已诱导表达了proteina的微生物菌液,总体积为50ml,其在波长为600nm的光照下的光吸收值od600为4.124,从v总中取出的用于检测目的蛋白表达情况的菌液体积为5ml。那么,取出的菌液经离心去上清液后,得到菌体沉淀,往菌体沉淀中加入的裂解缓冲液体积为824ul。
将所述体积为824ul的含菌体沉淀的裂解缓冲液混匀,并用超声破碎仪将菌体破碎后,各取30ul的破菌液于两个1.5mlep管中。其中一个ep1管中加入10ul的4*蛋白样品处理液,用于跑变性蛋白电泳的全菌液样品s全菌;另一个ep2管于高速离心机中离心,将离心后的上清液小心转入另一个新的ep3管中,并向ep3管中加入10ul的4*蛋白样品处理液,用于跑变性蛋白电泳的上清液样品s上清;已移除上清液的ep2管中所剩下的沉淀即为包涵体,往已移除上清液的ep2管中包涵体加入与上清液体积相同的裂解缓冲液30ul,并向ep2管中加入10ul的4*蛋白样品处理液,用于跑变性蛋白电泳的包涵体样品s包涵体。
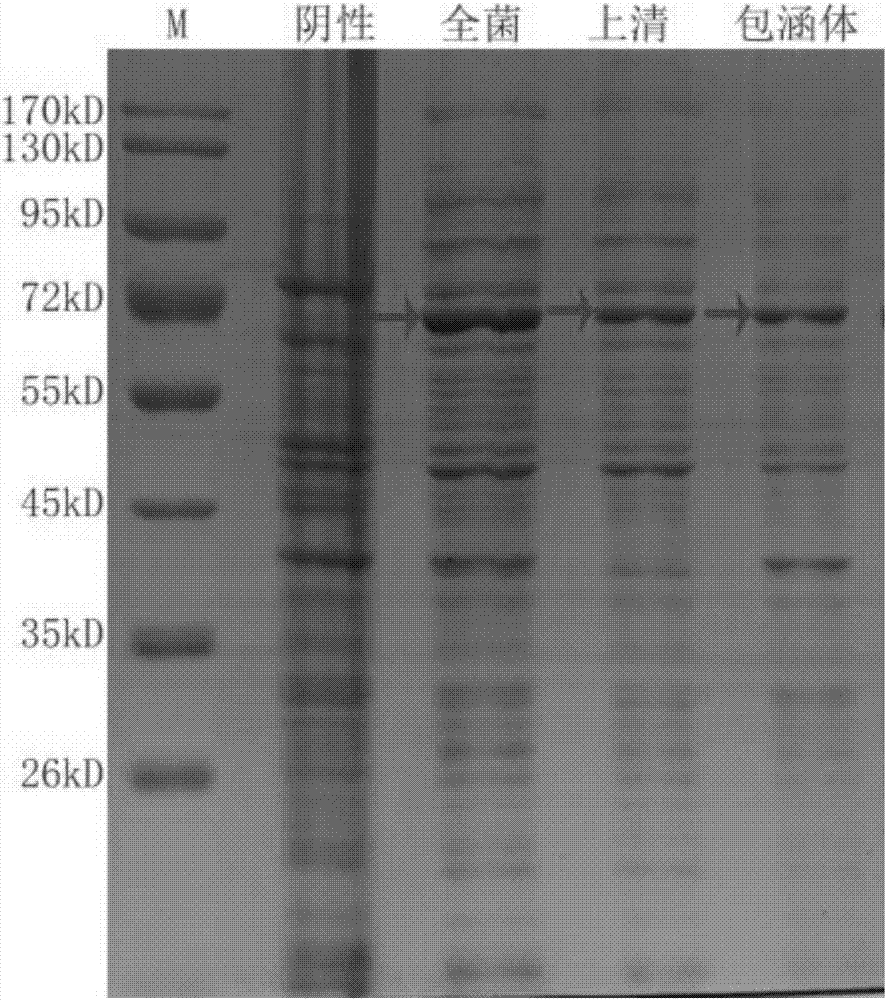
所述s全菌,s上清,s包涵体,按同样方法制得的不含目的蛋白的全菌液样品s阴性和已知蛋白浓度为1ug/ul的纯蛋白s定量连同蛋白maker一起跑变性蛋白电泳,除s定量上样量为1ul外,其余上样量都为15ul。蛋白电泳胶图结果如图1所示。
通过电泳胶图定性分析为:有实验组有目的蛋白表达,且上清液中目的蛋白比包涵体中的目的蛋白量更少些。
通过bio-radimagelab凝胶成像系统分析定量分析蛋白电泳胶图,可得出
s全菌:s定量=1.27,s上清:s定量=0.48,s包涵体:s定量=0.71,
从而得到上样量都为15ul的s全菌,s上清,s包涵体里目的蛋白的量分别为1.27ug,0.48ug,0.71ug,其中m全菌大致等于m上清+m包涵体。
因此,可溶性目的蛋白量与包涵体中目的蛋白总量的比值为d=m上清/m包涵体=0.676,表明可溶性蛋白表达量相比当包涵体少了32.4%。
从而体积为50ml的微生物菌液中可溶性目的蛋白的总量
m上清=160m上清v总a/(3v上样)ug=352ug;
体积为v总ml的微生物菌液中可溶性目的蛋白的浓度为
d上清=160m上清a/(3v上样)ug/ml=7ug/ml=7mg/l。表明在该表达条件下,每升培养基能表达出7mg的可溶性目的蛋白,这样的表达情况不算特别好,需要后续优化表达条件或优化质粒构建。
实验例
实验组:完全严格按实施例中公开的,本发明的方法来;
对照组1:与实验组相比,区别在于:没有加定量对照的纯蛋白;
对照组2:与实验组相比,区别在于:没有加阴性对照的样品和定量对照的纯蛋白;
对照组3:与实验组相比,区别在于:未按本发明的方法来,但是随意加的,未加定量蛋白。
对照组1
按上述方法处理样品,跑电泳,但是没有加定量对照的纯蛋白。电泳胶图结果如图2所示。
从电泳胶图可以看出,对比阴性对照组,实验组有目的蛋白表达,且上清液中目的蛋白量比包涵体中目的蛋白量多一些,表明可溶性目的蛋白表达量相对多一些。但因为没有加定量对照的纯蛋白,无法定量确定目的蛋白的绝对量,因此无法准确确定目的蛋白的表达水平。因些有必要加上定量蛋白。
对照组2
按上述方法处理样品,跑电泳,但是没有加阴性对照的样品和定量对照的纯蛋白。电泳胶图结果如图3所示。
因为没有阴性对照样品,所以无法确定哪条带是目的蛋白。因此更谈不上定性和定量分析。于是一定要记得加上阴性对照的样品。
对照组3
该实验例中,取菌液时未量体积,也未测菌液的od600,经离心得到菌体沉淀后,随意加了一定体积的裂解缓冲液。超声波破菌后,未按上述方法处理跑电泳前的样品,未算好体积加样品和蛋白样品处理液,只是随意处理样品,未加定量对照的纯蛋白,电泳结果如图4所示。
从电泳胶图可以看出,对比阴性对照组,实验组有目的蛋白表达。但因为处理样品时未算好体积,而是随意加样,导致包涵体中目的蛋白量比全菌中的目的蛋白还要多,因此不能反映目的蛋白在上清液和包涵体中的表达分布情况。即使假如加了定量蛋白,可能通过凝胶分析系统得出全菌、上清液和包涵体中目的蛋白的量,但因为没有记录制备电泳样品的各项数据,从而也是无法定量算出可溶性目的蛋白的表达水平。因此制备电泳样品时要遵循上述方法。
本发明技术效果主要体现在以下方面:本方法发明操作简单方便,所用到的仪器设备都是生物实验室常用仪器。从定量的角度上来分析蛋白表达情况,解决现有问题,为后续蛋白表达优化,蛋白放大生产及分离纯化提供可靠依据。
当然,以上只是本发明的典型实例,除此之外,本发明还可以有其它多种具体实施方式,凡采用等同替换或等效变换形成的技术方案,均落在本发明要求保护的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!