一种PEG及PEG化药物的分析检测方法及其应用与流程
本发明属于药物分析研究技术领域,涉及到分析peg及peg化药物生物质谱方法。
背景技术:
近年来,为适应药物研发的需要,药物分析科学和技术得到了长足的发展。近年来,迅速发展的液相色谱串联质谱技术(lc-ms/ms)为peg化药物分析提供了可能的解决方案。同传统的免疫学、hplc、比色法等相比,液相色谱串联质谱方法在准确度、精密度、选择性、灵敏度和定量动态范围等各方面均显示出较大优势。目前,对于peg化小分子药物的质谱定量分析报道较少,其主要原因是peg化的蛋白类药物可以采取蛋白质组学的方法选择酶解后的特征肽段进行定量分析;但对于peg化小分子药物尚无有效的酶切手段将其水解为游离的药物分子。另外,peg化药物的分子量分布范围较宽,即使相同聚合度的peg分子量也不唯一,而基于mrm等扫描模式的lc-ms/ms手段只能对有限的和分子量确定的目标化合物进行定量分析,因此,对多组分多形态的高分子peg化药物进行质谱分析存在着很大的挑战性。源内裂解技术在一级质谱中能给出丰富的离子碎片信息。利用解簇电压(declusteringpotential,dp)影响离子进入质谱的速度,锥孔的解簇电压越高,离子速度越快,离子损失越小,检测灵敏度越高。过高的锥孔电压会增加离子间的碰撞,引起源内裂解,产生碎片离子。这些碎片离子既可用于物质的定性,也可以用于mrm扫描模式同时监测母离子和碎片离子来实现定量分析。通过在离子源内加入较大能量解簇电压(dp)使peg及peg化药物发生源内裂解,生成特异性碎片离子,选择特异性的碎片离子进行监测,完成对peg及peg化药物的定量分析;但是,由于dp的能量较小,cidinsource的碰撞效率有所限制,该方法并不适合某些结合能力较强的peg化药物的分析,尤其是较难得到peg化药物针对药物的特定碎片离子。当键合到小分子(<500)上的peg分子量较大时(>10k)时,游离peg与peg化药物色谱行为非常接近,很难通过色谱分离,此时只是通过监测peg特异性的碎片来完成peg和peg化药物的监测就不合适了;另外,由于dp的碰撞能量较小,该方法的灵敏度也有所限制。massall技术能够在一定程度上提供peg及peg化药物的定量方法,但是由于massall技术是使所有的带电粒子全部通过q1进入碰撞室打碎成二级碎片,进入碰撞室的离子没有选择性,同时由于同一时刻进入碰撞室离子数目过多,会显著降低一级离子的裂解效率和传输效率,灵敏度和选择性均会受到限制,同时由于不能得到peg或peg化药物在不同质荷比范围内的色谱质谱峰响应分布情况,该方法应用于peg或peg化药物的定性分析能力有限。
技术实现要素:
本发明要解决的技术问题是,建立一种可以同时兼顾母离子扫描和母离子分段进入碰撞室发生碰撞诱导解离获得特定碎片离子的技术方案,能够获得高分子聚合物前体离子在特定质荷比区间的分布,根据母离子和碎片离子同时完成peg及peg化药物的定性及定量分析。本发明的目的是通过以下技术方案实现的:
一种peg及peg化药物的分析检测方法,使用液相色谱-四极杆-飞行时间质谱(lc-q-q-tofms)进行分析检测,将含有peg及peg化药物的待测物通过液相色谱分离,并建立tofms-swath连续可变窗口,分别设置tofms对应的碰撞能量和swath对应的碰撞能量;通过设定连续可变的窗口使不同质荷比范围的带电离子依次进入碰撞室q2,待测物经能量碰撞产生特异性碎片,通过对特异性碎片进行扫描分析对待测物进行检测。
进一步的,tofms对应的碰撞能量为10ev,swath对应的碰撞能量为40ev。
进一步的,swath进行质荷比采集窗口的通道依次为500-600,600-700,700-800,800-900,900-1000,1000-1100,1100-1200,1200-1250。
进一步的,选取的peg特异性碎片离子为atm/z89.0611,133.0869,177.1102,221.1366,265.1622,309.1878,353.2108,397.2359,即2个,3个,4个,5个,6个,7个,8个,9个peg单元。
进一步的,待测物中检测目标为peg550,其特定质荷比丰度最高的分布窗口为m/z500-700;待测物中检测目标为peg750,其特定质荷比丰度最高的分布窗口为m/z400-800;待测物中检测目标为peg2000,其特定质荷比丰度最高的分布窗口为m/z600-800;待测物中检测目标为peg5000,其特定质荷比丰度最高的分布窗口为m/z600-800;待测物中检测目标为peg2000-dox,其特定质荷比丰度最高的分布窗口为m/z600-1000。
一种peg及peg化阿霉素的分析检测方法,使用液相色谱-四极杆-飞行时间质谱仪(lc-q-q-tofms)进行分析检测,将含有peg及peg化药物的待测物通过液相色谱分离,并建立tofms-swath连续可变窗口,分别设置tofms对应的碰撞能量和swath对应的碰撞能量;通过设定连续可变的窗口使不同质荷比范围的带电离子依次进入碰撞室q2,待测物经能量碰撞产生特异性碎片,通过对特异性碎片进行扫描分析对待测物进行检测,其检测条件为:
色谱条件为:高效液相色谱系统;色谱柱:300sbc18柱,150mm×4.6mmi.d.,5μm粒径;流动相:含体积百分数占0.1%甲酸的水和含体积百分数占0.1%甲酸的乙腈,梯度洗脱;柱温30~40℃;流速0.8~1.0ml/min;进样量50μl;
质谱条件为:q-q-tof型串联质谱仪,配有esi离子化源和analyst数据处理软件;离子源:esi离子化源;正离子方式检测;离子喷射电压4500v;温度500℃;源内气体1:氮气压力50psi;气体2:氮气压力50psi;气帘气体:氮气压力25psi;tofms扫描模式,解簇电压(dp电压):80v;碰撞能量(ce电压):10ev;swath扫描方式,解簇电压(dp电压):80v;碰撞能量(ce电压):40ev,ces15ev;swath进行质荷比采集窗口的通道依次为500-600,600-700,700-800,800-900,900-1000,1000-1100,1100-1200,1200-1250。
进一步的,选取的peg特异性碎片离子atm/z89.0611,133.0869,177.1102,221.1366,265.1622,309.1878,353.2108,397.2359;阿霉素的特异性碎片离子atm/z321.0838,361.0785;peg化阿霉素的特异性碎片离子atm/z365.0735。
进一步的,测定生物样品中peg及peg化阿霉素在测定之前包括样品前处理和标准曲线制备步骤,取经由前处理后的上清液进行液相色谱-四极杆-飞行时间质谱分析,记录色谱图,将peg、阿霉素和peg化阿霉素峰面积代入标准曲线,求得peg、阿霉素和peg化阿霉素浓度。
本研究首次提出了基于连续可变窗口采集技术的cidin-quadrupole的lc-q/q/tof串联质谱方法,利用四级杆飞行时间质谱(tripletof5600)的高分辨率及高质量准确度,建立tofms-swath连续可变窗口技术,tofms对应的碰撞能量为10ev,swath对应的碰撞能量为40ev;通过设定连续可变的窗口使不同质荷比范围的带电离子依次进入碰撞室q2,分析物经能量碰撞产生特异性碎片,通过对特异性碎片进行扫描分析,建立peg及peg化药物稳定、可靠的lc/ms/ms分析方法。更重要的是,通过swath的采集窗口连续变化,可以在不同质荷比窗口内得到peg或peg化药物的色谱质谱峰,从而根据峰面积可以得出其在不同质荷比窗口的响应比值及分布。同时,由于窗口的连续分段采集,离子传输效率高,同一时刻进入碰撞室的离子数目不会超过碰撞室裂解能力的阈值,裂解效率更好,得到的二级碎片的信息丰度和灵敏度更好。综上,运用swath连续可变窗口采集技术,一次采集就可以同时获得高分辨率的一级母离子信息和二级碎片信息,可以同时完成peg及peg化药物的定性及定量分析。peg特异性的高分辨率的特定碎片离子(atm/z89.0611133.0869,177.1102,221.1366,265.1622,309.1878,353.2108,397.2359,2个,3个,4个,5个,6个,7个,8个,9个peg单元)及药物特定的离子(阿霉素的特定碎片离子atm/z321.0838,361.0785;peg化阿霉素的特定碎片离子atm/z365.0735;),通过监测peg特异性和药物特异性的碎片离子来完成peg化药物及游离peg的分析,该方法重现性好,灵敏度高,高分辨率的碎片离子选择能够有效的消除干扰离子的影响,适合复杂生物基质中peg及peg化药物的分析。
本发明的优势在于:
1)一次采集就可以依次通过tofmsscan和swath同时获得高分辨率的一级母离子信息和二级碎片信息的用于定性及定量分析peg及peg化药物的生物质谱方法。
2)通过swath的不同采集通道可以明确peg及peg化药物在不同质荷比范围的分布丰度,母离子通过swath分段进入碰撞室裂解,传输效率和裂解效率更高,灵敏度和选择性更好。
下面结合附图及具体实施方式对本发明作进一步详细说明。
附图说明
图1是串联质谱对peg化药物特异性碎片进行扫描流程图。
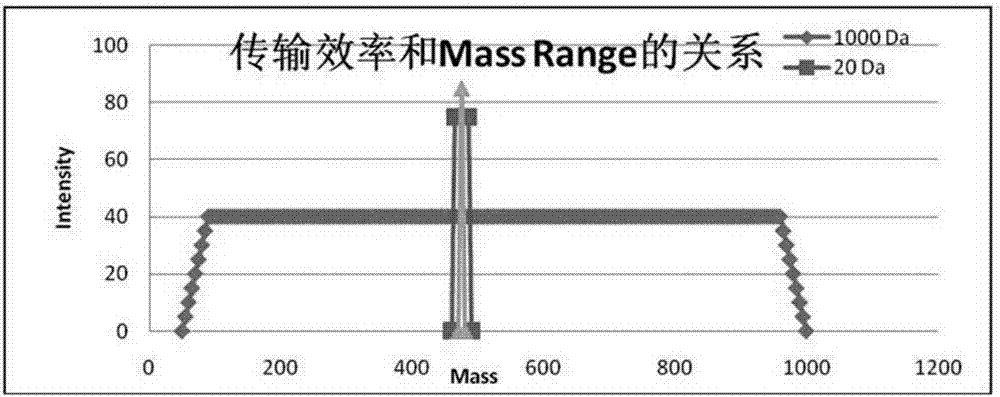
图2是传输效率与msrange的关系。
图3是实施例1得到的大鼠尾静脉给予peg化阿霉素后阿霉素及peg化阿霉素血药浓度时间曲线。
图4是实施例1得到的mpeg2000、阿霉素及peg化阿霉素典型色谱图(a,xicof133.08+/-0.15da;b,xicof321.063+/-0.008da;c,xicof361.063+/-0.008da;d,xicof365.057+/-0.008da)。
图5是实施例2得到的mpeg550扫描质谱图。
图6是实施例2得到的mpeg1000扫描质谱图。
图7是实施例2得到的mpeg550,mpeg750,mpeg2000,mpeg5000母离子质荷比分布图。
图8是实施例2得到的mpeg2k-阿霉素母离子质荷比分布图。
具体实施方式
参见图1、2,本发明的技术方案是一种可以一次采集就可以依次通过tofmsscan和swath同时获得高分辨率的一级母离子信息和二级碎片信息的用于定性及定量分析peg及peg化药物的生物质谱方法。本发明使用液相色谱-四极杆-飞行时间质谱(lc-q-q-tofms)进行分析检测,将含有peg及peg化药物的待测物通过液相色谱分离,并建立tofms-swath连续可变窗口,分别设置tofms对应的碰撞能量和swath对应的碰撞能量;通过设定连续可变的窗口使不同质荷比范围的带电离子依次进入碰撞室q2,待测物经能量碰撞产生特异性碎片,通过对特异性碎片进行扫描分析对待测物进行检测。
本发明可用于生物样品和非生物样品中的peg及多种peg化药物的检测。非常适合生物样品中peg及peg化阿霉素的检测,检测的具体步骤如下:
具体步骤如下:
a、生物样品处理:
1)于聚乙烯管中加入50μl生物样品,
2)加入50μl内标,涡流混匀,
3)加入200μl乙腈,涡流混匀,
4)12000rpm离心10分钟,上清液转移到聚乙烯管;
b、标准曲线制备:
1)利用乙腈-水(1/1,v/v)溶液将peg、阿霉素和peg化阿霉素储备液分别稀释至0.1、0.2、0.6、1.0、2.0、6.0、10.0μg/ml;
2)取50μl进行液相色谱-串联质谱分析,记录色谱图,以peg、阿霉素和peg化阿霉素浓度为横坐标,peg、阿霉素和peg化阿霉素峰面积为纵坐标,用加权w=1/x2最小二乘法进行回归运算,求得的直线回归方程,即为标准曲线;
c、生物样品中游离药物和peg化药物含量测定:
取经由a步骤处理后的上清液50μl进行液相色谱-串联质谱分析,记录色谱图,将peg、阿霉素和peg化阿霉素峰面积代入标准曲线,求得peg、阿霉素和peg化阿霉素浓度;所述的b步骤标准曲线制备及c步骤生物样品中阿霉素和peg化阿霉素含量测定。
色谱条件为:高效液相色谱系统;色谱柱:300sbc18柱,150mm×4.6mmi.d.,5μm粒径;流动相:含体积百分数占0.1%甲酸的水和含体积百分数占0.1%甲酸的乙腈,梯度洗脱;柱温30~40℃;流速0.8~1.0ml/min;进样量50μl;
质谱条件为:q-q-tof型串联质谱仪,配有esi离子化源和analyst数据处理软件;离子源:esi离子化源;正离子方式检测;离子喷射电压4500v;温度500℃;源内气体1:氮气压力50psi;气体2:氮气压力50psi;气帘气体:氮气压力25psi;tofms扫描模式,解簇电压(dp电压):80v;碰撞能量(ce电压):10ev;swath扫描方式,解簇电压(dp电压):80v;碰撞能量(ce电压):40ev,ces15ev;swath进行质荷比采集窗口的通道依次为500-600,600-700,700-800,800-900,900-1000,1000-1100,1100-1200,1200-1250;
本发明的peg及peg化药物生物质谱分析方法中,所述的色谱条件中的梯度洗脱,程序见下表,
其中a是含体积百分数占0.1%甲酸的水,b是含体积百分数占0.1%甲酸的乙腈。
本发明的peg化小分子药物生物质谱绝对定量方法,还可以在样品测定期间利用质量控制样品对方法进行验证;所述的质量控制样品,按照如下步骤制备:
1)利用乙腈-水(1/1,v/v)溶液将peg、阿霉素和peg化阿霉素储备液分别稀释至0.2、1.0、6.0μg/ml;
2)peg、阿霉素和peg化阿霉素每浓度取三个样本,根据标准曲线,得出阿霉素和peg化阿霉素浓度,计算质量控制样品准确度。
下面通过两个具体的实施例进行详细介绍。
实施例1
将载药量等同于1mg阿霉素的peg化阿霉素用1ml生理盐水溶解,大鼠尾静脉给药,采血时间点如下:0、0.05、0.083、0.167、0.25、0.5、1、1.5、2、3、4、6、12、24、48、72h。测定大鼠尾静脉给予peg化阿霉素之后血浆中游离阿霉素和peg化阿霉素的含量,血浆药物浓度时间曲线见图3。
主要步骤如下:
a、血浆样品预处理
1)于聚乙烯管中加入50μl血浆样品,
2)加入50μl内标,涡流混匀,
3)加入200μl乙腈,涡流混匀,
4)12000rpm离心10分钟,上清液转移到聚乙烯管;
b、标准曲线制备
1)利用乙腈-水(1/1,v/v)溶液将peg、阿霉素和peg化阿霉素储备液分别稀释至0.1、0.2、0.6、1.0、2.0、6.0、10.0μg/ml;
2)取50μl进行液相色谱-串联质谱分析,记录色谱图,以peg、阿霉素和peg化阿霉素浓度为横坐标,peg、阿霉素和peg化阿霉素峰面积为纵坐标,用加权w=1/x2最小二乘法进行回归运算,求得的直线回归方程,即为标准曲线;
如表1所示。
表1peg化阿霉素生物质谱绝对定量方法典型标准曲线
c、质量控制样品制备
按“标准曲线制备”项下操作,制备低、中、高三个浓度(0.2、1.0、6.0μg/ml)质量控制样品,每浓度至少三样本,根据标准曲线,得出浓度。
计算质量控制样品准确度,具体见表2,考察方法准确性。
表2peg化阿霉素生物质谱绝对定量方法质量控制样品准确度
上述步骤中涉及peg化阿霉素生物质谱绝对定量方法测定的条件如下:
色谱条件为:高效液相液相色谱系统;色谱柱:300sbc18柱,150mm×4.6mmi.d.,5μm粒径;流动相:含体积百分数占0.1%甲酸的水和含体积百分数占0.1%甲酸的乙腈,梯度洗脱;柱温40℃;流速1ml/min;进样量50μl;
所述的色谱条件中的梯度洗脱,程序见表3,
表3梯度洗脱程序
其中a是含体积百分数占0.1%甲酸的水,b是含体积百分数占0.1%甲酸的乙腈。
质谱条件为:q-q-tof型串联质谱仪,配有esi离子化源和analyst数据处理软件;离子源:esi离子化源;正离子方式检测;离子喷射电压4500v;温度500℃;源内气体1:氮气压力50psi;气体2:氮气压力50psi;气帘气体:氮气压力25psi;tofms扫描模式,解簇电压(dp电压):80v;碰撞能量(ce电压):10ev;swath扫描方式,解簇电压(dp电压):80v;碰撞能量(ce电压):40ev,ces15ev;swath进行质荷比采集窗口的通道依次为500-600,600-700,700-800,800-900,900-1000,1000-1100,1100-1200,1200-1250;
鉴于peg2k,阿霉素和peg化阿霉素典型标准曲线的线性(表1)以及质量控制样品的准确度(表2),本发明所述的一种同时测定peg,peg化阿霉素和游离阿霉素的生物质谱绝对定量方法线性关系良好、准确度高、重现性好而且灵敏、可靠,可用于上述peg,peg化药物及游离药物的绝对定量。
实施例2
利用乙腈-水(1/1,v/v)溶液将peg和peg化阿霉素储备液分别稀释至1.0μg/ml;取50μl进行液相色谱-串联质谱tofms-swath分析,记录质谱图及色谱图,对不同质荷比通道内药物的特异性碎片进行定量,得出peg及peg化药物在不同质荷比范围内的分布,可用于不同分子量的peg及peg化药物的定性分析。
色谱条件为:高效液相液相色谱系统;色谱柱:300sbc18柱,150mm×4.6mmi.d.,5μm粒径;流动相:含体积百分数占0.1%甲酸的水和含体积百分数占0.1%甲酸的乙腈,梯度洗脱;柱温30℃;流速0.8ml/min;进样量50μl;
所述的色谱条件中的梯度洗脱,程序见表4,
表4梯度洗脱程序
其中a是含体积百分数占0.1%甲酸的水,b是含体积百分数占0.1%甲酸的乙腈。
质谱条件为:q-q-tof型串联质谱仪,配有esi离子化源和analyst数据处理软件;离子源:esi离子化源;正离子方式检测;离子喷射电压4500v;温度500℃;源内气体1:氮气压力50psi;气体2:氮气压力50psi;气帘气体:氮气压力25psi;tofms扫描模式,解簇电压(dp电压):80v;碰撞能量(ce电压):10ev;swath扫描方式,解簇电压(dp电压):80v;碰撞能量(ce电压):40ev,ces15ev;swath进行质荷比采集窗口的通道依次为500-600,600-700,700-800,800-900,900-1000,1000-1100,1100-1200,1200-1250。
- 还没有人留言评论。精彩留言会获得点赞!