一种水中痕量N-亚硝胺的检测方法与流程
本发明属于环境检测与分析领域,尤其涉及一种水中痕量n-亚硝胺的检测方法。
背景技术:
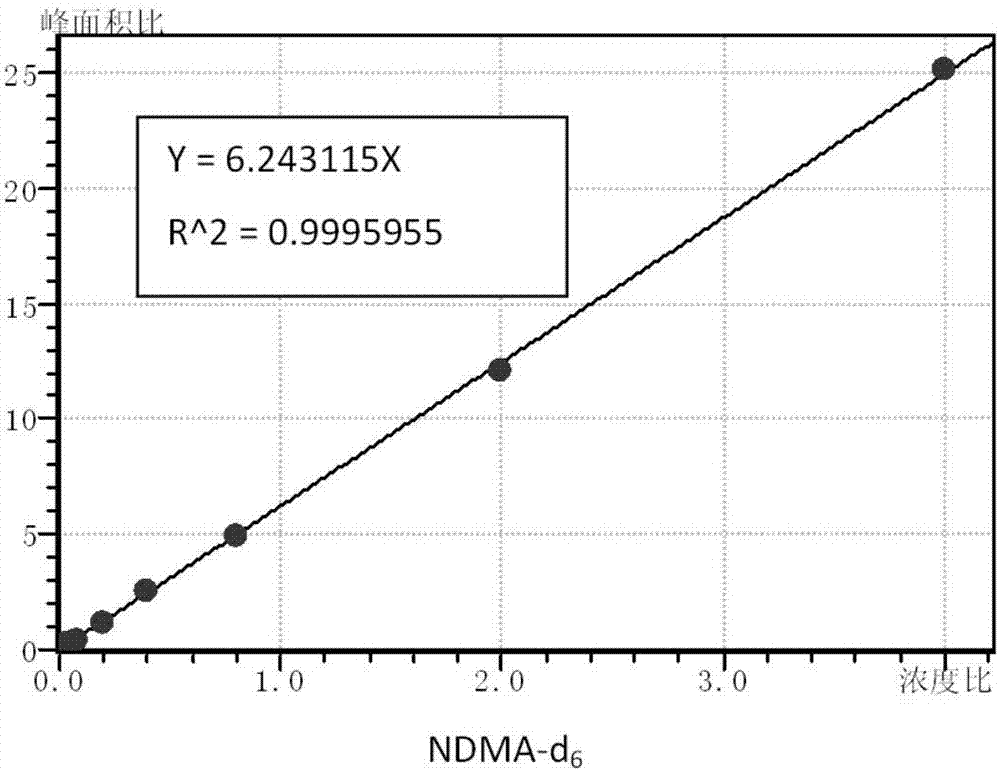
:n-亚硝胺是有潜在致癌性的一类化合物。由于前体化合物如亚硝酸盐和有机氮在水中进行亚硝化或氧化反应可以形成n-亚硝胺,因此这类化合物在国外自来水出厂水和管网末梢水中具有较高的检出率,引起了人们的广泛关注。由于n-亚硝胺有较高的辛醇/水分配系数,且其常以痕量浓度存在于水体当中,因此检出较为困难。目前,我国还没有统一的检测水中痕量n-亚硝胺的方法,所以探究一种检测限低、操作方便、测定成本较低的水中痕量n-亚硝胺的检测方法,是极为迫切的。技术实现要素:有鉴于此,本发明的目的在于提供一种水中痕量n-亚硝胺的检测方法,本发明提供的方法检测限低,操作方便,成本低。本发明提供了一种水中痕量n-亚硝胺的检测方法,包括以下步骤:a)、将待测水样与n-亚硝基二甲胺-d6混合后通过活性炭固相萃取柱,之后将所述活性炭固相萃取柱中的水分挤出,再用二氯甲烷对所述活性炭固相萃取柱进行洗脱,收集洗脱液;b)、将所述洗脱液与甲醇混合,进行浓缩,得到浓缩液;将所述浓缩液与n-亚硝基二丙基胺-d14混合,得到待分析样品;c)、对所述待分析样品进行气相色谱-质谱检测,根据检测结果和预先建立的标准曲线计算出所述待测水样中n-亚硝胺的种类和含量。优选的,步骤a)中,所述待测水样与n-亚硝基二甲胺-d6的用量比为1l:(80~150)ng。优选的,步骤a)中,所述待测水样与二氯甲烷的体积比为1l:(10~20)ml。优选的,步骤a)中,所述活性炭固相萃取柱的填料为椰壳活性炭。优选的,步骤a)中,所述活性炭固相萃取柱在通过待测水样与n-亚硝基二甲胺-d6的混合液之前,先对所述活性炭固相萃取柱进行活化。优选的,步骤a)中所述待测水样与步骤b)中所述甲醇的体积比为1l:(20~80)μl。优选的,步骤b)中,在所述洗脱液与甲醇混合之前,先对所述洗脱液进行干燥。优选的,步骤b)中,所述浓缩具体包括:所述洗脱液与甲醇混合后依次进行旋转蒸发和挥发,得到浓缩液;所述旋转蒸发的温度为15~35℃。优选的,步骤a)中所述待测水样与步骤b)中所述待分析样品的体积比为1l:(0.2~0.8)ml。优选的,步骤b)中,所述待分析样品中n-亚硝基二丙基胺-d14的含量为0.2~0.3mg/l。与现有技术相比,本发明提供了一种水中痕量n-亚硝胺的检测方法。本发明提供的方法包括以下步骤:a)、将待测水样与n-亚硝基二甲胺-d6混合后通过活性炭固相萃取柱,之后将所述活性炭固相萃取柱中的水分挤出,再用二氯甲烷对所述活性炭固相萃取柱进行洗脱,收集洗脱液;b)、将所述洗脱液与甲醇混合,进行浓缩,得到浓缩液;将所述浓缩液与n-亚硝基二丙基胺-d14混合,得到待分析样品;c)、对所述待分析样品进行气相色谱-质谱检测,根据检测结果和预先建立的标准曲线计算出所述待测水样中n-亚硝胺的种类和含量。本发明利用活性炭固相萃取和浓缩对水样进行预处理,可将原始水样浓缩数千倍,达到gc-ms的最低检测限;同时本发明通过结合使用同位素稀释法和gc-ms分析法,既可对水样中多种痕量n-亚硝胺进行定量测定,又可直观评价检测方法的稳定性。本发明提供的方法在水样预处理时步骤简便,容易操作;在样品分析时所用的gc-ms设备价格相对低廉,且在大多数分析实验室都已配备,因此该方法具有较好的推广前景。实验结果表明:采用本发明提供的方法对水样中的n-亚硝胺进行检测时,n-亚硝胺的检测限为2~6ng/l,回收率大约在60~80%左右。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。图1是本发明实施例1提供的2.0mg/ln-亚硝胺标准溶液的质谱峰图;图2是本发明实施例1提供的ndma-d6的标准曲线图;图3是本发明实施例1提供的mdma的标准曲线图;图4是本发明实施例1提供的nmea的标准曲线图;图5是本发明实施例1提供的ndea的标准曲线图;图6是本发明实施例1提供的npyr的标准曲线图;图7是本发明实施例1提供的nmor的标准曲线图;图8是本发明实施例1提供的ndpa的标准曲线图;图9是本发明实施例1提供的npip的标准曲线图;图10是本发明实施例1提供的ndba的标准曲线图;图11是本发明实施例1提供的ndpha的标准曲线图。具体实施方式下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。本发明提供了一种水中痕量n-亚硝胺的检测方法,包括以下步骤:a)、将待测水样与n-亚硝基二甲胺-d6混合后通过活性炭固相萃取柱,之后将所述活性炭固相萃取柱中的水分挤出,再用二氯甲烷对所述活性炭固相萃取柱进行洗脱,收集洗脱液;b)、将所述洗脱液与甲醇混合,进行浓缩,得到浓缩液;将所述浓缩液与n-亚硝基二甲胺-d14混合,得到待分析样品;c)、对所述待分析样品进行气相色谱-质谱检测,根据检测结果和预先建立的标准曲线计算出所述待测水样中n-亚硝胺的种类和含量。在本发明中,首先在待测水样中加入内标物,并对其进行固相萃取。其具体操作为:先将待测水样与n-亚硝基二甲胺-d6混合后通过活性炭固相萃取柱。其中,所述待测水样中优选含有一定量的n-亚硝胺,所述n-亚硝胺包括但不限于n-亚硝基二甲胺(ndma)、n-亚硝基甲基乙基胺(nmea)、n-亚硝基二乙基胺(ndea)、n-亚硝基二丙基胺(ndpa)、n-亚硝基二丁基胺(ndba)、n-亚硝基吡咯烷(npyr)、n-亚硝基吗啉(nmor)、n-亚硝基哌啶(npip)和n-亚硝基二苯胺(ndpha)中的一种或多种;所述内标物为n-亚硝基二甲胺-d6(ndma-d6);所述待测水样与n-亚硝基二甲胺-d6的用量比优选为1l:(80~150)ng,更优选为1l:100ng。在本发明中,使用活性炭固相萃取柱进行固相萃取,所述活性炭固相萃取柱的填料为椰壳活性炭;所述活性炭固相萃取柱的填料填充量优选为0.3~0.4g/ml。在本发明提供的一个实施例中,所述活性炭固相萃取柱为6ml的固相萃取小柱,装填2g的椰壳活性炭。在本发明中,优选在使用所述活性炭固相萃取柱进行固相萃取之前,先对其进行活化,所述活化的步骤包括:依次将二氯甲烷、甲醇和水通过活性炭固相萃取柱。其中,所述二氯甲烷优选分2~4次通入,更优选分2次通入,每次通入量优选为2~6ml,更优选为3ml;所述甲醇优选分2~6次通入,更优选份4次通入,每次通入量优选为2~6ml,更优选为3ml;所述水优选分3~7次通入,更优选分5次通入,每次通入量优选为2~6ml,更优选为3ml。在本发明中,待测水样与n-亚硝基二甲胺-d6的混合液通过活性炭固相萃取柱的流速优选为5~20ml/min,更优选为10ml/min;待测水样与n-亚硝基二甲胺-d6的混合液通过活性炭固相萃取柱的时间优选为50~200min,更优选为100min。在待测水样与n-亚硝基二甲胺-d6的混合液通过活性炭固相萃取柱的过程中,混合液中的n-亚硝胺不断富集到活性炭上,待混合液通入结束后,将所述活性炭固相萃取柱中的水分挤出,再用二氯甲烷对所述活性炭固相萃取柱进行洗脱,将其上富集的n-亚硝胺洗脱下来。其中,所述二氯甲烷与所述待测水样的体积比优选为(10~20)ml:1l,更优选为15ml:1l。洗脱完毕后,收集洗脱液。得到洗脱液后,对所述洗脱液进行浓缩。在本发明中,为去除洗脱液中可能残留的水,优选在对洗脱液进行浓缩前,先对所述洗脱液进行干燥。所述干燥使用的干燥剂优选为无水硫酸钠,所述无水硫酸钠与所述待测水样的用量比优选为(3~8)g:1l,更优选为6g:1l。在本发明中,优选在使用无水硫酸钠对洗脱液进行干燥前,先对所述污水硫酸钠进行活化和清洗。其中,所述活化的温度优选为400~500℃,更优选为450℃;所述活化的时间优选为1~3h,更优选为2h;所述清洗使用的清洗剂优选为二氯甲烷;所述清洗剂与无水硫酸钠的用量比优选为(10~15)ml:(3~8)g,更优选为12ml:6g;所述清洗剂优选分4次对无水硫酸钠进行清洗。在本发明中,为了降低干燥对对检测结果的影响,优选在洗脱液干燥完毕后对干燥剂进行淋洗,并将淋洗得到的流出液合并入干燥后的洗脱液中。其中,所述淋洗所用的淋洗液优选为二氯甲烷;所述淋洗液与无水硫酸钠的用量比优选为(4~10)ml:(3~8)g,更优选为6ml:6g;所述淋洗液优选分2次对无水硫酸钠进行淋洗。在本发明中,为减少在洗脱液浓缩时n-亚硝胺的损失,在浓缩前先将所述洗脱液与甲醇混合。其中,所述甲烷与所述待测水样的体积比优选(20~80)μl:1l,更优选为50μl:1l。待所述洗脱液与甲醇混合后,进行浓缩,所述浓缩优选包括:依次进行旋转蒸发和挥发。其中,所述旋转蒸发的温度优选为15~35℃,更优选为25℃;本发明对所述旋转蒸发的温度没有特别限定,蒸发至需求体积即可;旋转蒸发得到的液体与所述待测水样的体积比优选为(0.3~0.8)ml:1l,更优选为0.5ml:1l。在本发明中,旋转蒸发完毕后,加入一定量的二氯甲烷冲洗旋转蒸发的容器内壁,所述二氯甲烷与所述旋转蒸发得到的液体的体积比优选为1:(0.3~1),更优选为1:0.5。冲洗容器内壁后,将容器内容物移至棕色样品瓶中,之后挥发到一定体积,得到浓缩液。在本发明中,所述浓缩液与所述待测水样的体积比优选<(0.3~0.8)ml:1l,更优选<0.5ml:1l。得到浓缩液后,将所述浓缩液与n-亚硝基二丙基胺-d14混合,定容,得到待分析样品。其中,所述待分析样品中n-亚硝基二丙基胺-d14的含量优选为0.2~0.3mg/l,更优选为0.25mg/l;所述待分析样品与所述待测水样的体积比优选为(0.2~0.8)ml:1l,更优选为0.5ml:1l。得到待分析样品后,对所述待分析样品进行气相色谱-质谱(gc-ms)检测,优选进行气相色谱-电子轰击离子源质谱(gc-ei/ms)检测。在本发明中,检测所用的气相色谱-质谱检测仪优选为岛津gcms-qp2010ultra,色谱柱pat-5:30m×0.25mmi.d.×0.5μm。检测过程中,电子能量优选为-60~-80ev,更优选为-70ev;载气优选为氦气,进行方式优选为不分流进样;进样口温度优选为240~260℃,更优选为250℃;传输线温度优选为270~290℃,更优选为280℃;离子源温度优选为200~220℃,更优选为210℃;载气流速优选为1~3ml/min,更优选为2.0ml/min;程序升温条件优选为:初始柱温设为30~40℃,优选为35℃,保持2~6min,优选为4min;之后升温至210~230℃,优选升温至220℃,升温速率为6~10℃/min,优选为8℃/min;再升温至270~290℃,优选升温至280℃,保持1~3min,优选为2min,升温速率为30~50℃/min,更优选为40℃/min。检测完毕后,得到gc-ms检测结果,根据检测结果和预先建立的标准曲线计算出所述待测水样中n-亚硝胺的种类和含量。在本发明提供的一个实施例中,可以按照以下方式建立标准曲线:i)配制一系列不同种类n-亚硝胺的标准溶液,所述n-亚硝胺的种类包括但不限于n-亚硝基二甲胺(ndma)、n-亚硝基甲基乙基胺(nmea)、n-亚硝基二乙基胺(ndea)、n-亚硝基二丙基胺(ndpa)、n-亚硝基二丁基胺(ndba)、n-亚硝基吡咯烷(npyr)、n-亚硝基吗啉(nmor)、n-亚硝基哌啶(npip)和n-亚硝基二苯胺(ndpha);所述标准溶液的浓度梯度优选为0.01、0.02、0.05、0.10、0.20、0.50、1.00、2.00mg/l;所述标准溶液中添加ndpa-d14作为内标,ndpa-d14添加量与处理待测水样时的添加量一致。ii)对配制的一系列标准溶液分别进行气相色谱-质谱检测,其具体检测过程和检测条件的选择与上文检测待测水样时一致。iii)检测结束后,得到不同种类、不同浓度n-亚硝胺标准溶液对应的峰面积和内标ndpa-d14的峰面积,针对每种n-亚硝胺,以n-亚硝胺与内标ndpa-d14的峰面积比值为横坐标,以n-亚硝胺与内标ndpa-d14的浓度比值为纵坐标,绘制标准曲线。本发明利用活性炭固相萃取和浓缩对水样进行预处理,可将原始水样浓缩数千倍,达到gc-ms的最低检测限;同时本发明通过结合使用同位素稀释法和gc-ms分析法,既可对水样中多种痕量n-亚硝胺进行定量测定,又可直观评价检测方法的稳定性。本发明提供的方法在水样预处理时步骤简便,容易操作;在样品分析时所用的gc-ms设备价格相对低廉,且在大多数分析实验室都已配备,因此该方法具有较好的推广前景。实验结果表明:采用本发明提供的方法对水样中的n-亚硝胺进行检测时,n-亚硝胺的检测限为2~6ng/l,回收率大约在60~80%左右。为更清楚起见,下面通过以下实施例进行详细说明。下述实施例中,使用的gc-ms分析仪为岛津gcms-qp2010ultra,色谱柱pat-5:30m×0.25mmi.d.×0.5μm;使用的固相萃取小柱为6ml的固相萃取小柱,装填2g的椰壳活性炭。实施例11)gc-ms测试条件采用gc-ms分析9种n-亚硝胺的标准溶液(2.0mg/l),离子源为ei源,电子能量为-70ev,载气为高纯氦气,不分流进样1l。实验的进样口温度250℃,传输线温度280℃、离子源温度210℃,载气流速为2.0ml/min。n-亚硝胺的gc/ms程序升温条件:初始柱温设为35℃,保持4min,以8℃/min的速度升至220℃,再以40℃/min的速度升至280℃,保持2min。采用保留时间和特征碎片离子定性,选择离子检测法(sim)和内标法绘制标准曲线进行定量分析,不同n-亚硝胺的gc-ms分析参数如表1所示,不同n-亚硝胺的标准溶液的质谱峰图如图1所示,图1是本发明实施例1提供的2.0mg/ln-亚硝胺标准溶液的质谱峰图。表1n-亚硝胺的gc-ms分析参数2)标准曲线绘制2-1)配制标准溶液:配制浓度梯度为0.01、0.02、0.05、0.10、0.20、0.50、1.00、2.00mg/l的亚硝胺标准溶液,内标ndpa-d14含量为0.25mg/l,保存在4℃冰箱中,待测。2-2)gc-ms检测和建立标准曲线:在步骤1)的测试条件下对步骤2-1)获得的待测样品进行检测,得到检测结果。以n-亚硝胺与内标ndpa-d14的峰面积比值为横坐标,以n-亚硝胺与内标ndpa-d14的浓度比值为纵坐标,作标准曲线,结果如图2~11所示。图2~11分别为本发明实施例1提供的ndma-d6、mdma、nmea、ndea、npyr、nmor、ndpa、npip、ndba、ndpha的标准曲线图。由图2~11可以看出,9种亚硝胺标准曲线的相关性系数扩值均在0.996-0.999左右,峰面积和浓度的相关性较好,可满足后续分析测试的要求。3)样品预处理:3-1)固相萃取小柱的活化:依次将试剂通过固相萃取柱:3ml二氯甲烷×2次;3ml甲醇×4次;3ml超纯水×5次。3-2)水样的富集:将1l水样以10ml/min的流速通过固相萃取柱,可采取水样自流,不外加真空泵抽气。3-3)洗脱:用注射器活塞将固相萃取柱中的水分缓慢挤出,将15ml二氯甲烷缓慢通过固相萃取柱,洗脱活性炭上富集的亚硝胺物质,收集洗脱液。3-4)干燥:收集到的洗脱液经6g无水硫酸钠(无水硫酸钠需在450℃活化两小时后再用3ml二氯甲烷×4次清洗湿润)干燥,再用3ml二氯甲烷×2次淋洗无水硫酸钠,收集流出液。3-5)浓缩:在干燥后的洗脱液和收集的流出液中加入50μl甲醇,在室温(25℃)下采用旋转蒸发浓缩至0.5ml左右,加入1ml左右二氯甲烷冲洗内壁,移至棕色样品瓶中自然挥发到一定体积,加入一定量ndpa-d14,定容到0.5ml,内标ndpa-d14含量为0.25mg/l,保存在4℃冰箱中,待测。4)检出限的测定与计算:采用标准偏差(sd)法测定n-亚硝胺的检出限(dl):用超纯水配制7份浓度为10ng/l的n-亚硝胺溶液,按照步骤3-1)~3-5)的方法对配制的溶液进行处理,得到待测样品,在步骤1)的测试条件下对待测样品进行检测,根据检测结果和对应的标准曲线计算出样品中n-亚硝胺的含量,算出各n-亚硝胺溶液的平均浓度和浓度的标准偏差(sd),dl的计算公式为:dl=3.143×sd。其中,3.143为平行样份数为7时,t(n-1,1-a=0.99)的值。结果如表2所示:表2n-亚硝胺的检测限n-亚硝胺sd(ng/l)dl(ng/l)ndma1.34.09nmea1.23.77ndea1.65.03npyr1.13.46nmor0.92.83ndpa1.34.09npip1.13.46ndba1.65.03ndpha1.85.66通过表2可以看出,本实施例提供的检测方法的检测限为2-6ng/l。实施例2模拟水样中9种亚硝胺测定使用supelco公司的9种n-亚硝胺混标(2000mg/l甲醇溶液)和ndma-d6(1000mg/l二氯甲烷)配制混合水溶液(n-亚硝胺总含量0.5μg/l,ndma-d6含量100ng/l)1l,按照实施例1步骤3-1)~3-5)的方法对水样进行处理,得到待测水样,在实施例1步骤1)的测试条件下对待测水样进行检测,根据检测结果和对应的标准曲线计算出模拟水样中各n-亚硝胺的含量和回收率。设置三组平行实验,9种亚硝和ndma-d6的回收率实验结果如表3所示:表39种亚硝和ndma-d6的回收率通过表3可以看出,除去最后一种ndpha回收率较低在20%左右,nmea与ndba回收率在55%左右,其余的都有70-85%左右。同时,通过表3可以看出,ndma-d6的回收率为60%-80%。在实际样品预处理前,通过向水样中加入100ng/l的ndma-d6作为内标,可评价样品预处理方法的稳定性,再根据仪器对ndma-d6的响应值估算出测试方法中物质的回收率范围。实施例3模拟前驱物氯化亚硝胺的测定尼扎替丁(胃药)确认为亚硝胺前驱物之一。使用去离子水配制1l浓度为24μg/l尼扎替丁溶液。在25℃,溶液ph=8条件下,1l尼扎替丁溶液与2ml浓度为1.8g/l的一氯胺溶液在暗处氯化反应24h之后加入淬灭剂,添加ndma-d6作为内标(添加量100ng/l),之后按照实施例1步骤3-1)~3-5)的方法对其进行预处理,得到待测水样,在实施例1步骤1)的测试条件下对待测水样进行检测,根据检测结果和对应的标准曲线计算出水样中n-二甲基亚硝胺(ndma)的含量、ndma-d6的回收率和尼扎替丁的摩尔转化率,设置三组平行实验,结果如表4所示:表424μg/l尼扎替丁氯化亚硝胺的测定结果表4中,换算后得到的水样实际ndma按照以下公式计算:上式中,c换算为换算后得到的水样实际n-亚硝胺浓度,单位μg/l;c测定为预处理后水样进行gc-ms测定后的响应值对应的标准曲线上的n-亚硝胺浓度,即仪器测定的n-亚硝胺浓度,单位mg/l;v1为进行预处理的水样体积,单位ml,本次计算中为1000;v2为预处理后的水样体积,单位ml,本次计算中为0.5;p为ndma-d6的回收率,即ndma-d6在gc-ms中测定的浓度与实际浓度的比值。通过表4可以发现,ndma-d6的回收率在60%左右,水样中的ndma浓度在0.27μg/l左右,其摩尔转化率与文献shenr,andrewssa.demonstrationof20pharmaceuticalsandpersonalcareproducts(ppcps)asnitrosamineprecursorsduringchloraminedisinfection[j].waterresearch,2011,45(2):944-952.的结果相符合。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1