本发明涉及一种双酚类化合物的分子印迹固相萃取-高效液相色谱-串联质谱测定方法和应用,属于环境有机污染物分离分析领域。
背景技术:
双酚a(bpa)是目前使用最广泛的化合物之一,全球的年产量在80亿英镑左右。bpa是聚碳酸酯、环氧树脂和酚醛树脂生产过程中的原料或添加剂,也是外科修补术中抗氧化和稳定化处理的重要材料,在食品包装和容器内壁涂装中也被广泛使用。材料的生产、制造、使用过程中的溶出都有可能释放出bpa,并通过大气或污水进入环境。研究表明bpa在环境,食品和人体样品中广泛存在,美国90%的普通人尿样中都检出了bpa。bpa具有基因诱变性和雌激素干扰作用,微量甚至痕量的浓度也可能对动物生理状况、生殖系统以及胎儿发育等造成不良影响,是一种环境内分泌干扰物(edcs)。bpa已经被美国和欧盟一些国家和地区作为重点列入优先控制污染物的黑名单。伴随法规对于bpa使用的限制,双酚s(bps)、双酚f(bpf)、双酚e(bpe)、双酚b(bpb)、双酚af(bpaf)、双酚ap(bpap)和双酚z(bpz)等作为bpa的主要替代物被越来越多的使用,其毒性和雌激素效应与双酚a相似。介于双酚类对于人体的潜在毒性,对环境中双酚类物质含量的评估是必要的,可以为风险控制提供可靠的参考数据。
低浓度和基质复杂是限制双酚类物质分析检测的瓶颈。双酚类在环境中的存在浓度很低,一般都在ngl-1或ngg-1级,需要较高的样品富集倍数和高灵敏的质谱检测。其中,固相萃取-高效液相色谱-串联质谱检测(spe-hplc-ms/ms)是近年来发展的最有潜力的双酚类化合物检测方法。常用于双酚类富集的固相萃取材料有hlb、c18和mcx等,但均存在选择性低、杂质去除能力差的缺点,未去除的样品基质和杂质会在液相色谱分离过程中与双酚类物质共馏出,一起进入电喷雾电离源,进而影响双酚类化合物的电离效率,表现为电离增强或抑制效应,影响分析方法的准确性和重现性。
近年来发展的替代模板分子印迹固相萃取(dmispe)技术,可以在保留分子印迹材料对目标分子识别选择性的同时,有效地避免其因模板泄漏而造成的分析结果不准确问题,具有很高的选择性和可靠性。本发明所使用的双酚类化合物分子印迹固相萃取材料采用1,1,1-三(4-羟基苯基)乙烷(thpe)作为替代模板,对双酚类化合物具有超高的类选择性(zl201310334888.8)。将替代模板分子印迹固相萃取与lc-ms/ms相结合可以同时获得低的基质效应和高的检测灵敏度,克服现有技术的不足。本发明采用双酚类化合物替代模板印迹材料作为固相萃取填料结合hplc-ms/ms建立环境样品中9种双酚类内分泌干扰物的高效分离检测方法,具有非常高的科研和实用价值。
技术实现要素:
针对现有技术不足,本发明提供一种双酚类化合物替代模板分子印迹固相萃取-高效液相色谱-串联质谱测定方法用于环境样品中痕量双酚类内分泌干扰物的检测。
为实现上述目的,本发明公开了一种双酚类化合物分子印迹固相萃取-高效液相色谱-串联质谱测定方法,所述方法的测定步骤如下:
(1)样品的采集与保存
采集的水样放置于棕色玻璃瓶中,污泥或底泥样品置于不锈钢饭盒中。样品采集后,在现场加入甲醛(1%,v/v)去除生物活性,冷藏运输。样品运抵实验室后,水样采用玻璃纤维滤膜过滤,并在-20℃保存待用。污泥、底泥或土壤样品经冷冻干燥后,过60目筛,并于-20℃保存待用。
(2)水样的富集和净化
水样采用氢氧化钠调节至ph9.0,加入bpa-13c12和tbbpa-13c12同位素内标后加载至双酚类化合物专用的分子印迹固相萃取柱管。分子印迹固相萃取柱预先采用3ml乙腈和3ml水活化。水样的上样量为10-1000ml,完成上样后,对固相萃取柱真空抽干10-30min。然后采用3ml乙腈淋洗,最后用12.0ml甲醇或6ml甲醇-三氟乙酸(98:2,v/v)洗脱。洗脱液采用氮吹浓缩至近干,并采用甲醇-水(50:50,v/v)定容至1ml。定容后样品用于hplc-ms/ms分析,进样量为10μl。
(3)污泥、底泥或土壤样品的提取与净化
0.2-10g污泥、底泥或土壤样品(干重)加入bpa-13c12和tbbpa-13c12同位素内标后,采用有机溶剂提取三次。提取过程为超声5-30min外加涡旋5-30min,然后3000-6000g下离心5-30min,取上清液。合并三次的提取液于12ml的玻璃管中,在氮吹下浓缩至约4ml,加入超纯水至10ml用于分子印迹固相萃取上样。将10ml提取液以1mlmin-1的速度加载至双酚类化合物专用的固相萃取柱管,然后采用3ml水淋洗并抽干10-30min。抽干后采用3.0ml乙腈淋洗,12.0ml甲醇或6ml甲醇-三氟乙酸(98:2,v/v)洗脱。洗脱液采用氮吹浓缩至近干,并采用甲醇-水(50:50,v/v)定容至1ml。定容后样品用于hplc-ms/ms分析,进样量为10μl。
(4)样品分析
使用液相色谱-串联质谱进行分析。
高效液相色谱条件:采用c18高效液相色谱柱,甲醇和水为流动相,柱温30℃,进样量10μl,流动相流速为200μlmin-1。测定时的流动相梯度如下:35%-100%甲醇(25min),100%甲醇(5min),100%-35%(2min),35%甲醇(5min)。
三重四级杆串联质谱条件:采用电喷雾(esi)离子源,负电离模式,多反应离子监测(multiplereactionmonitoring,mrm)扫描定量分析目标污染物。毛细管电喷雾电压为-2500v,离子源温度和溶剂蒸发温度均为300℃,鞘气和辅助气(99%纯度)压力分别为5和20psi,离子扫描时间为0.2s,扫描宽度为0.5m/z。
步骤(1)中所用玻璃纤维滤膜的孔径为0.45μm;
步骤(1)和(2)中所述水样可以是自来水、饮用水、地表水或污水等。
步骤(2)和(3)中所述双酚类化合物专用分子印迹固相萃取柱的规格为200mg,6ml。
步骤(3)中用于污泥、底泥或土壤样品提取的机溶剂为甲醇、乙腈、丙酮、或甲醇-水混合溶液;
步骤(3)和(4)中氮吹浓缩时的加热温度为30-50℃;
步骤(4)中c18高效液相色谱柱的规格为(150×2.1mm;5μm)。
本发明的双酚类化合物分子印迹固相萃取-高效液相色谱-串联质谱测定方法具有较高的灵敏度、准确度和精密度,可应用于环境、食品和生物等复杂样品中痕量双酚类内分泌干扰物的分析检测。
附图说明
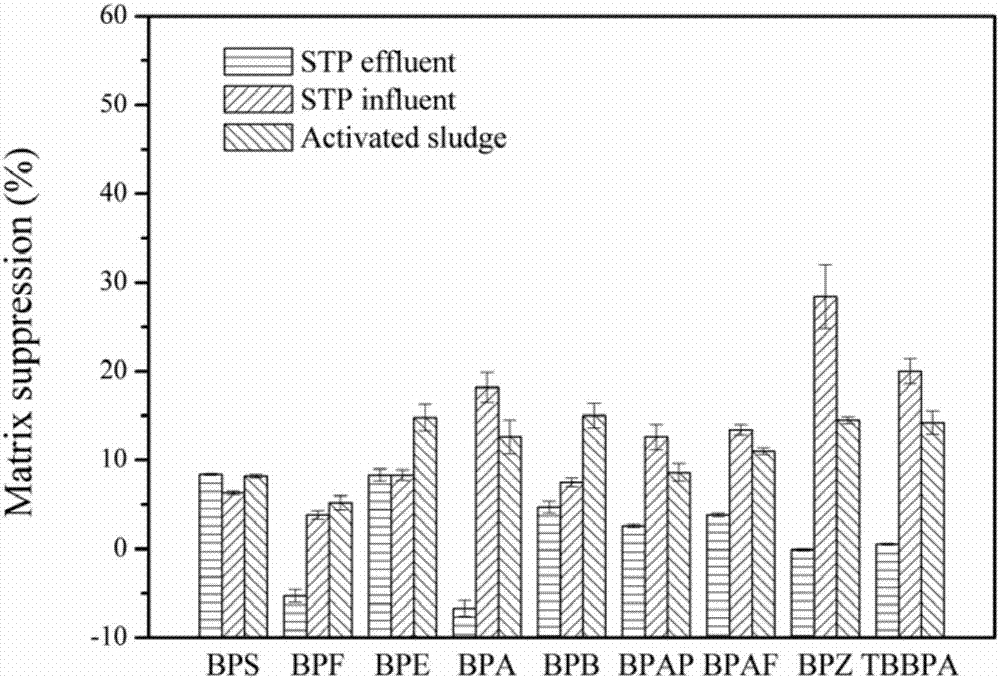
图1.不同洗脱条件对基质效应的影响。
图2.进口污水、出口污水和污泥中双酚类化合物的基质效应。
图3.溶剂组成对污泥中双酚类化合物提取效率的影响。
图4.双酚类化合物分子印迹固相萃取柱与商业化固相萃取柱净化效果对比。
具体实施方式
下面对本发明的优选实施例进行详细阐述,以使本发明的优点和特征能更易于被本领域技术人员理解,从而对本发明的保护范围做出更为清楚明确的界定。
实施例
污水和污泥中9种双酚类化合物分子印迹固相萃取-高效液相色谱-串联质谱测定方法包括以下部分:
1.实验部分
1.1分子印迹固相萃取柱的制备
1mmol模板分子(thpe)溶于5.6ml乙腈,混匀后依次加入功能单体(4mmol)、交联剂edma(20mmol)和引发剂40mg,制得预聚合溶液。将预聚合溶液经超声混匀后置于冰浴中,通氮气10min后放置于4℃冷藏2h,后取出置于60℃水浴中反应24h。制备得到的块状聚合物经破碎、研磨、筛分得到粒径在38.5-63μm的颗粒物,丙酮沉降除去小颗粒物。采用甲醇-乙酸(90:10,v/v)为溶剂索氏抽提48h去除模板分子和未反应的单体、交联剂等,然后于60℃真空干燥过夜。获得的印迹材料用于自制固相萃取柱填料,每根3ml的固相萃取空柱管手动装填200mg该分子印迹填料。
1.2样品的采集与保存
采集的污水样品放置于棕色玻璃瓶中,污泥样品置于不锈钢饭盒中。样品采集后,在现场加入甲醛(1%,v/v)去除生物活性,冷藏运输。样品运抵实验室后,水样采用玻璃纤维滤膜过滤,并在-20℃保存待用。污泥样品冷冻干燥后,过60目筛,并于-20℃保存待用。
1.3污水样品的富集和净化
水样采用氢氧化钠调节至ph9.0,加入bpa-13c12和tbbpa-13c12同位素内标后加载至双酚类化合物专用的分子印迹固相萃取柱管。分子印迹固相萃取柱预先采用3ml乙腈和3ml水活化。水样的上样量为10-1000ml,完成上样后,对固相萃取柱真空抽干30min。然后采用3ml乙腈淋洗,最后用12.0ml甲醇洗脱或6ml甲醇-三氟乙酸(98:2,v/v)洗脱。洗脱采用氮吹浓缩至近干,并采用甲醇-水(50:50,v/v)定容至1ml。定容后样品用于hplc-ms/ms分析,进样量为10μl。
1.4污泥样品的提取与净化
0.2g污泥样品(干重)加入bpa-13c12和tbbpa-13c12同位素内标后,采用有机溶剂提取三次。所用溶剂为甲醇-水(5:3,v/v)、甲醇-丙酮(1:1,v/v)、甲醇-水(8:2,v/v)或甲醇-水(ph=12.0)(5:3,v/v)。提取过程为超声5min外加涡旋30min,然后4500g下离心5min,取上清液。合并三次的提取液于12ml的玻璃管中,在氮吹下浓缩至约4ml,加入超纯水至10ml用于分子印迹固相萃取上样。将10ml污泥提取液以1mlmin-1的速度加载至双酚类化合物专用的固相萃取柱管,然后采用3ml水淋洗并抽干30min。抽干后采用3.0ml乙腈淋洗,12.0ml甲醇洗脱。洗脱采用氮吹浓缩至近干,并采用甲醇-水(50:50,v/v)定容至1ml。定容后样品用于hplc-ms/ms分析,进样量为10μl。
1.5仪器条件
使用液相色谱-串联质谱(hplc-ms/ms)进行分析。
高效液相色谱条件:采用c18高效液相色谱柱,甲醇和水为流动相,柱温30℃,进样量10μl,流动相流速为200μlmin-1。测定时的流动相梯度如下:35%-100%甲醇(25min),100%甲醇(5min),100%-35%(2min),35%甲醇(5min)。
三重四级杆串联质谱条件:采用电喷雾(esi)离子源,负电离模式,多反应离子监测(multiplereactionmonitoring,mrm)扫描定量分析目标污染物。毛细管电喷雾电压为-2500v,离子源温度和溶剂蒸发温度均为300℃,鞘气和辅助气(99%纯度)压力分别为5和20psi,离子扫描时间为0.2s,扫描宽度为0.5m/z。各化合物的母离子、子离子、碰撞能和透镜电压见表1。
表1hplc-ms/ms质谱参数
在上述规定的色谱、质谱条件下,分别以m/z108.1、93.3、198.0、212.0、224.2、212.0、274.0、264.9、222.9、445.7、296.7为bps、bpf、bpe、bpa、bpa-13c12、bpb、bpap、bpaf、bpz、tbbpa、tbbpa-13c12的定量离子,以bpa-13c12和tbbpa-13c12为同位素内标,采用内标曲线法进行定量。
1.6方法的质量控制
痕量双酚类化合物质谱检测的准确性可能会受到很多因素的影响,在整个分析过程中,应严格的做好质量控制。本实验中,首先对溶剂和程序空白进行了考察。对spe和hplc过程中使用的甲醇和乙腈进行了浓缩和检测,结果表明没有检出任何双酚类物质。进一步对程序空白进行了测定(每批样品都跟一个程序空白),检出了包括bps、bpa、tbbpa和bpaf在内的四种双酚,浓度水平分别为0.01、0.01、0.21和0.02ngml-1。其次,在分析过程中,每隔10针样品插入一针20ppb双酚混标,以考察仪器的稳定性。最后,每10针样品插入一针溶剂空白(甲醇),以考察进样过程中样品间的交叉污染。结果表明bpaf存在一定程度的交叉污染,将进样的洗针溶剂改为乙腈-异丙醇(50:50,v/v)后,bpaf的交叉污染降至0.0002ngml-1。
1.7方法验证
为确保双酚类化合物检测的准确性和可靠性,对方法的线性范围、检出限、回收率、精密度以及基质效应进行了系统考察。将1000ppm双酚混标储备液用流动相稀释制成1ppm作为母液,通过逐级稀释的方法配置了从0.00005-100ppb范围内的13个浓度点作为系列标准溶液。每个标准溶液中都加入20ngtbbpa-13c12同位素内标,采用内标曲线进行双酚类物质定量,所得内标定量曲线线性均在0.99以上。
采用加标回收的方法考察了回收率和精密度。实验中对300ml污水和0.2g活性污泥样品进行了加标回收实验,加标浓度为20ppb。在此基础上,对方法的日内和日间精密度进行了考察。日内精密度测定的方法为:五份加标样品,当天同一批处理,当天完成测定。日间精密度的测定方法为:五份加标样品,每天处理和测定一个样品,连续5天完成所有测定。三种不同基质均完成上述精密度测定。本实验中,因为找不到完全空白的样品作为加标基质,回收率计算需扣除样品本底。
按照美国临床实验室标准委员会(nccls)的定义,基质效应是指样品中除分析物以外的其它成分对分析物测定值的影响。lc-ms/ms中的基质效应由分析物的共馏出组分影响电喷雾接口的离子化效率所致,表现为电离增强或抑制效应。在lc-ms/ms过程中,基质效应会严重影响方法的精密度和准确度。基质效应按测定方法的不同,可以分为绝对基质效应和相对基质效应。本实验按照matuszewski等提出的方案对绝对基质效应进行了考察。具体测定方法如下:制备两组待测样品,一组为用纯溶剂配置的20ppb双酚类标样,另一组为采用经固相萃取、浓缩复溶后的样品空白基质配置的20ppb双酚类标样,对这两组样品分别进行lc-ms/ms测定,采用公式me(%)=(1-am/a0)进行计算。式中,me(%)代表绝对基质效应,若me(%)为正值,存在基质抑制,若me(%)为负值,则存在基质增强。am为样品空白基质中双酚类的测定值,a0为标样中双酚类的测定值。方法的检出限和定量限分别定义为3倍和10倍信噪比时的检测浓度。
2.结果与讨论
2.1分子印迹固相萃取淋洗和洗脱条件优化
采用10ml100ng的bps混配纯水溶液上样,发现当淋洗体积为4.0ml时,包括tbbpa在内的9种双酚的回收率均大于85%,进一步增加淋洗体积会导致tbbpa回收率的降低。由于污水和污泥样品基质复杂、含量高,为了避免在上样和淋洗过程中由于杂质占据吸附位点而导致回收率降低,同时也为了提高方法对于不同基质的适用性,将乙腈淋洗体积定为3.0ml。
将3.0ml乙腈淋洗和6.0ml甲醇-三氟乙酸(98:2,v/v)洗脱用于100ml进口污水(ph=9.0)的富集净化,并对基质效应进行了考察,发现基质抑制较严重。这可能是由于,污水基质中含有较多酸性物质,乙腈淋洗过程中不能有效的去除,并且会在甲醇-三氟乙酸(98:2,v/v)洗脱过程中洗脱下来,在双酚检测过程中,这些杂质能够与双酚共洗脱,并且影响其电离效率。为了降低这种基质抑制作用,对洗脱条件进行了重新优化。按照上述假设,在乙腈淋洗过程中不能除去的主要是酸性较强的物质,因此为了降低洗脱步骤对这些物质的洗脱强度,将洗脱液改为纯甲醇。实验表明,当采用12.0ml甲醇洗脱时,双酚类的回收率均大于85%,能够满足分析要求。进一步考察了12.0ml甲醇洗脱时的基质效应,并与6.0ml甲醇-三氟乙酸(98:2,v/v)洗脱条件下的基质效应进行了对比,结果如图1所示。从图中可以看出,采用12.0ml甲醇洗脱时基质抑制显著低于6.0ml甲醇-三氟乙酸(98:2,v/v)洗脱,因此选用12.0ml甲醇作为洗脱液。
2.2实际样品基质效应考察
采用上述2.1所述优化结果,即3.0ml乙腈淋洗,12.0ml甲醇洗脱,对进、出口污水和活性污泥样品中双酚类物质测定的基质效应进行了考察,结果如图2所示。进、出口污水和污泥中双酚类化合物的基质效应分别为3.8-28.4%、-6.7-8.4%和5.2-15.0%。出口污水和污泥样品没有表现出显著的基质效应,进口污水中的bpz基质抑制略高,达到28.4%。
2.3污泥样品提取优化
分别采用甲醇-水(5:3,v/v)、甲醇-丙酮(1:1,v/v)、甲醇-水(8:2,v/v)和甲醇-水(ph=12.0)(5:3,v/v)为提取溶剂,考察污泥中双酚类化合物的提取效率,结果如图3所示,甲醇-水(ph=12.0)(5:3,v/v)为最佳提取溶剂。
2.4与商业化固相萃取材料对比
双酚类化合物分子印迹固相萃取柱与商业化固相萃取柱样品净化效果对比如图1所示,采用双酚类化合物分子印迹固相萃取柱处理的样品基质效在-6.7-28.4%之间,远低于商业化固相萃取柱。
2.5方法性能
污水和污泥中9种双酚类化合物分子印迹固相萃取-高效液相色谱-串联质谱测定方法回收率、精密度和定量限考察数据如表2所示。结果表明,mispe-hplc-ms/ms方法对污水和污泥样品的加标回收率在43.6-101.2%之间,精密度在1.5-14.9%之间,具有较好的重复性和可靠性,能够用于实际样品的测定。
表2mispe-hplc-ms/ms方法的回收率、精密度和定量限
intra-pre:日内精密度
rec.:回收率
mloq:方法定量限
2.6实际样品测定
采用上述建立的mispe-lc-ms/ms方法对污水处理厂的四个污水和一个污泥样品进行了测定,结果列于表3中。由表中可以看出,除了bpb和bpz,其他双酚在污水中均有检出。其中bpa、bps、bpf和bpe的浓度较高,这与文献报道的bps和bpf为bpa主要替代物的报道符合。
表3污水和污泥样品的测定结果