同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法与流程
本发明属于市政给排水和环境工程的技术领域,涉及水质检测分析技术,具体涉及一种同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法。
背景技术:
饮用水消毒工艺通过灭活病原菌以达到保证饮用水安全的目的,但同时也因为产生了一系列消毒副产物,对人们的健康产生了威胁[1]。流行病学研究指出,人们长期饮用氯化消毒后的水可能会导致膀胱癌患病率的升高[2]。然而,研究人员并没有甄别出导致膀胱癌致病的消毒副产物[3]。另外,流行病学研究表明消毒副产物还可能危害人类生殖健康[4]。迄今为止,已报道的消毒副产物超过700种[5],但有50%以上的总有机卤素尚未查明[1],而这些未查明的消毒副产物很可能是潜在的致癌物质。因此,消毒副产物的检测甄别尤为重要。近年来,含氮消毒副产物(亚硝胺、卤化氰、卤乙腈、卤代乙酰胺和硝基甲烷等)由于其毒性高而备受关注[6]。随着检测技术的进步和检测精度的提高,饮用水中不断检测到芳香族消毒副产物。2010年,研究人员首次在实验水样中检测到了卤代苯类醌消毒副产物(hbqs)[7]。数据表明,hbqs广泛存在加拿大和美国的饮用水系统中,浓度水平为3.3-274ng/l[8],随后饮用水中检测到卤代苯甲酸、卤代苯酚和卤代硝基苯酚等芳香族消毒副产物的存在[1]。毒理学结果显示芳香族消毒副产物的细胞毒性比脂肪族消毒副产物高出数个数量级[2]。
含氮有机物和芳香类有机物在水中广泛存在[9],在消毒过程中,与氯/氯氨发生化学反应便产生了含氮芳香性消毒副产物。迄今,国内很少有同时针对饮用水中多种含氮芳香性消毒副产物的系统检测技术,氯苯乙腈是一类新型含氮芳香性消毒副产物,而氯苯乙腈的研究和检测方法从未被报道,因此,建立便捷、有效和快速的检测方法对6种氯苯乙腈的检测和控制研究至关重要。另外,大部分消毒副产物的检测使用气相色谱-电子捕获检测器,依靠标准物质的保留时间比较的方式定性,虽然可以较好地完成定性定量分析,但是对氯化反应中产生的中间产物则没有辨别能力,不便于开展消毒副产物生成机理的研究。
参考文献:
1.krasner,s.w.;weinberg,h.s.;richardson,s.d.;pastor,s.j.;chinn,r.;sclimenti,m.j.;
onstad,g.d.;thruston,a.d.,occurrenceofanewgenerationofdisinfectionbyproducts.environ.sci.technol.2006,40,(23),7175-7185.
2.villanueva,c.m.;cantor,k.p.;grimalt,j.o.;malats,n.;silverman,d.;tardon,a.;
garcia-closas,r.;serra,c.;carrato,a.;
epidemiol.2007,165,(2),148.
3.li,x.f.;mitch,w.a.,drinkingwaterdisinfectionbyproducts(dbps)andhumanhealtheffects:multidisciplinarychallengesandopportunities.environ.sci.269technol.2017.
4.grellier,j.;bennett,j.;patelarou,e.;smith,r.b.;toledano,m.b.;rushton,l.;briggs,d.j.;
nieuwenhuijsen,m.j.,exposuretodisinfectionby-products,fetalgrowth,andprematurity:asystematicreviewandmeta-analysis.epidemiology2010,21,(3),300-13.
5.richardson,s.d.;ternes,t.a.,wateranalysis:emergingcontaminantsandcurrentissues.anal.chem.2018,90,(1),398-428.
6.krasner,s.w.;mitch,w.a.;westerhoff,p.;dotson,a.,formationandcontrolofemergingc-andn-dbpsindrinkingwater.j.am.waterworksassn.2012,104,(11),e582-e595.
7.zhao,y.l.;qin,f.;boyd,j.m.;anichina,j.;li,x.f.,characterizationanddeterminationof
chloro-andbromo-benzoquinonesasnewchlorinationdisinfectionbyproductsindrinkingwater.anal.chem.2010,82,(11),4599-605.
8.zhao,y.l.;anichina,j.;lu,x.f.;bull,r.j.;krasner,s.w.;hrudey,s.e.;li,x.f.,
occurrenceandformationofchloro-andbromo-benzoquinonesduringdrinkingwaterdisinfection.waterres.2012,46,(14),4351-4360.
9.saunders,j.f.;yu,y.;mccutchanjr,j.h.;rosario-ortiz,f.l.,characterizinglimitsof
precisionfordissolvedorganicnitrogencalculations.2017.
技术实现要素:
本发明针对现有技术中的不足,目的是提供一种同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法。
为达到上述目的,本发明的解决方案是:
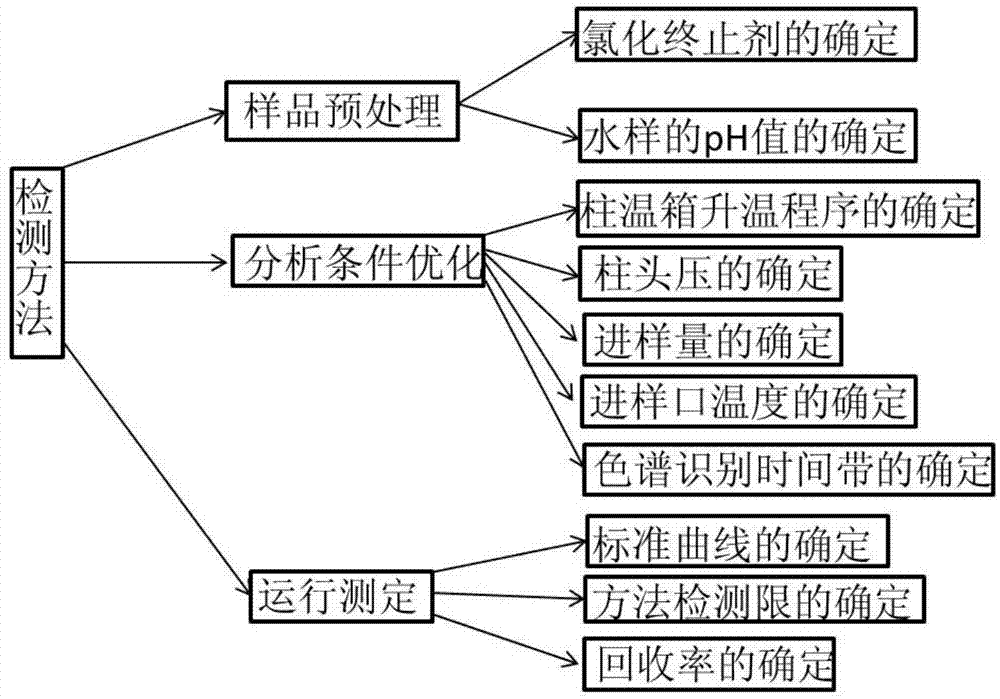
一种同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法,其包括:样品预处理、分析条件优化以及运行测定。
实际上,饮用水的水样中含氮芳香性消毒副产物氯苯乙腈由4-氯苯乙腈、2,3-二氯苯乙腈、2,4-二氯苯乙腈、2,5-二氯苯乙腈、2,6-二氯苯乙腈和3,4-二氯苯乙腈组成。
氯苯乙腈因氯原子数目和氯取代位置的差异,一共分为6种;其中,2,3-二氯苯乙腈、2,4-二氯苯乙腈、2,5-二氯苯乙腈、2,6-二氯苯乙腈和3,4-二氯苯乙腈为同分异构体。
进一步地,同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法具体包括如下步骤:
(1)、将饮用水的水样通过液液萃取预处理的方式进行富集;
(2)、确定气相色谱/质谱(gc/ms)联用仪的运行参数,包括柱温箱升温程序、柱头压、进样量、进样口温度和色谱识别时间带;
(3)、建立6种氯苯乙腈的标准曲线;
(4)、采用气相色谱/质谱联用仪分析该水样,根据得到的色谱图和质谱图进行定性分析,然后采用外标法建立的6种氯苯乙腈的标准曲线确定水样中6种氯苯乙腈的出峰时间。
在步骤(1)中,饮用水的水样进行富集的预处理的过程为:取10ml饮用水的水样置于25ml玻璃小瓶中,加入200μmol/l的终止剂脱氯后,调节该水样的ph值,加入2.0g无水硫酸钠,手动振荡使其充分溶解;再加入2.0ml萃取剂,使用ika振荡器在2500r/min下振荡5.0min,静置10.0min。
具体地,样品预处理的过程具体包括水样的ph值和氯化终止剂的确定。
进一步地,水样的ph值为5-9,水样的ph值优选为7。
进一步地,终止剂(即氯化终止剂)选自抗坏血酸、硫代硫酸钠、氯化铵、亚砷酸钠和亚硫酸钠中的一种以上。
进一步地,萃取剂为甲基叔丁基醚。
具体地,在步骤(2)中,采用气相色谱-质谱(gc/ms)联用仪进行分析检测,即分析条件优化,包括柱温箱升温程序、柱头压、进样量、进样口温度和色谱识别时间带的确定。
进一步地,柱头压为134.2±0.3kpa,优选为134.2kpa。
进一步地,进样量为1.0-10μl,优选为1.0μl。
进一步地,进样口温度为100-250.0℃,优选为230.0℃。
进一步地,色谱识别时间带为0.1-1.0min,优选为0.2min。
实际上,在气相色谱/质谱(gc/ms)联用仪中,每一种物质在气相色谱(gc)上都对应一个保留时间,在质谱(ms)上对应具有特征的离子碎片,因此,gc/ms联用仪的定性原理就是结合上述两者进行双重定性。上述分析参数(即柱温箱升温程序、柱头压、进样量、进样口温度和色谱识别时间带)的选择便决定了各种氯苯乙腈在gc上的保留时间,对各个分析参数进行优化的目的是为了使各种氯苯乙腈在gc上能够更好地分离;各种氯苯乙腈在ms中ei电离源(70ev电子)轰击下产生离子碎片。
具体地,运行测定包括标准曲线、方法检测限和回收率的确定。
在步骤(3)中,建立6种氯苯乙腈的标准曲线的具体过程为:
(a)、配置混合标准溶液:分别称取20mg的4-氯苯乙腈、20mg的2,3-氯苯乙腈、20mg的2,4-二氯苯乙腈、20mg的2,5-二氯苯乙腈、20mg的2,6-二氯苯乙腈和20mg的3,4-二氯苯乙腈,溶于20ml甲基叔丁基醚中,混合配置成各个氯苯乙腈浓度相同(初始浓度均为1mg/ml)的混合标准溶液,并置于棕色瓶中;
(b)、用超纯水(millipore,电阻率为18mω·cm)将步骤(a)中的混合标准溶液配置成梯度标准校正液,梯度质量浓度分别为1μg/l、5μg/l、10μg/l、20μg/l、50μg/l、100μg/l和200μg/l;
(c)、采用气相色谱/质谱联用仪对上述梯度标准校正液进行批处理分析,利用labsolution工作站分别生成6种氯苯乙腈的标准曲线,其中横坐标为氯苯乙腈的质量浓度,纵坐标为氯苯乙腈的峰面积。
采用本发明的检测方法,使得6种氯苯乙腈在分析过程中可实现良好分离,在1-200μg/l范围内线性良好(r^2>0.99),6种氯苯乙腈的回收率在87-95%之间;方法检测限(limitofmethoddetection,mdl)在100ng/l以下;相对标准偏差(relativestandarddeviation,rsd)小于10.0%。
更进一步地,在步骤(4)中,4-氯苯乙腈的出峰时间为11.110±0.005min,2,4-二氯苯乙腈的出峰时间为11.680±0.005min,2,5-二氯苯乙腈的出峰时间为13.150±0.005min,2,6-二氯苯乙腈的出峰时间为13.375±0.005min,2,3-二氯苯乙腈的出峰时间为13.600±0.005min,3,4-二氯苯乙腈的出峰时间为14.385±0.005min。
由于采用上述方案,本发明的有益效果是:
第一,本发明提供了同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的新思路,不同于现有技术单次检测单种消毒副产物的方法,大大提高了分析效率,从而为多种芳香性消毒副产物的同时测定奠定了基础。
第二,本发明首次提出了同时检测饮用水中新型含氮芳香性消毒副产物-氯苯乙腈,并采用gc/ms联用仪,通过对保留时间和离子碎片信息的双重定性,使得定性能力强和精准度高,进一步确定了gc/ms联用仪的分析参数,因此,其可同时实现6种氯苯乙腈的良好分离,避免出现峰拖尾等现象,保证其正常出峰并缩短了分析时间,从而得到了较高的方法检测限和较小的相对标准偏差。
第三,本发明的gc/ms联用仪采用自动进样,并确定了自动进样的运行参数,方便快捷,从而提高了效率;另外,本发明的gc/ms联用仪可利用样品中物质的离子碎片信息(即质荷比),与nist标准谱库中的离子碎片信息进行比较,从而识别分析中间产物的分子结构,为氯苯乙腈的生成机制研究提供了基础。
第四,本发明采用了小体积液液萃取的预处理方法,即提取和富集水样中的6种氯苯乙腈,在保证富集倍数的同时,相比以往的萃取方式(水样量为200ml),其节省了水样、氯化终止剂和萃取剂的消耗量,同时也降低了成本。
附图说明
图1为本发明的同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法的流程示意图。
图2为本发明的同时检测饮用水中含氮芳香性消毒副产物中4-氯苯乙腈的标准曲线示意图。
图3为本发明的同时检测饮用水中含氮芳香性消毒副产物中2,4-二氯苯乙腈的标准曲线示意图。
图4为本发明的同时检测饮用水中含氮芳香性消毒副产物中2,5-二氯苯乙腈的标准曲线示意图。
图5为本发明的同时检测饮用水中含氮芳香性消毒副产物中2,6-二氯苯乙腈的标准曲线示意图。
图6为本发明的同时检测饮用水中含氮芳香性消毒副产物中2,3-二氯苯乙腈的标准曲线示意图。
图7为本发明的同时检测饮用水中含氮芳香性消毒副产物中3,4-二氯苯乙腈的标准曲线示意图。
图8为本发明的同时检测饮用水中含氮芳香性消毒副产物中6种氯苯乙腈在全扫模式下的色谱图。
具体实施方式
本发明提供了一种同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法。
<同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法>
本发明的同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法包括:样品预处理、分析条件优化以及运行测定。
实际上,饮用水的水样中含氮芳香性消毒副产物氯苯乙腈由4-氯苯乙腈、2,3-二氯苯乙腈、2,4-二氯苯乙腈、2,5-二氯苯乙腈、2,6-二氯苯乙腈和3,4-二氯苯乙腈组成。
氯苯乙腈因氯原子数目和氯取代位置的差异,一共分为6种;其中,2,3-二氯苯乙腈、2,4-二氯苯乙腈、2,5-二氯苯乙腈、2,6-二氯苯乙腈和3,4-二氯苯乙腈为同分异构体。分子结构式具体如下:
4-氯苯乙腈的分子结构式为:
2,3-二氯苯乙腈的分子结构式为:
2,4-二氯苯乙腈的分子结构式为:
2,5-二氯苯乙腈的分子结构式为:
2,6-二氯苯乙腈的分子结构式为:
3,4-二氯苯乙腈的分子结构式为:
进一步地,同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法具体包括如下步骤:
(1)、将饮用水的水样通过液液萃取预处理的方式进行富集;
(2)、确定气相色谱/质谱(gc/ms)联用仪的运行参数,包括柱温箱升温程序、柱头压、进样量、进样口温度和色谱识别时间带;
(3)、建立6种氯苯乙腈的标准曲线;
(4)、采用气相色谱/质谱联用仪分析水样,根据得到的色谱图和质谱图进行定性分析,然后采用外标法建立的6种氯苯乙腈的标准曲线确定水样中6种氯苯乙腈的出峰时间。
[样品预处理]
在步骤(1)中,饮用水的水样进行富集的预处理的具体过程为:
取10ml饮用水的水样置于25ml玻璃小瓶中,加入200μmol/l的氯化终止剂脱氯后,调节该水样的ph值,加入2.0g无水硫酸钠,手动振荡使其充分溶解;再加入2.0ml萃取剂,使用ika振荡器在2500r/min下振荡5.0min,静置10.0min。
其中,加入无水硫酸钠的目的为:减少萃取过程中乳浊液的产生,从而提高萃取效率。
实际上,样品预处理包括水样的ph值和最佳氯化终止剂的确定。
(水样的ph值的确定)
进一步地,饮用水的水样进行萃取过程前需要调节ph值。
最佳ph值的确定过程为:测定6种氯苯乙腈分别在ph=5、ph=6、ph=7、ph=8和ph=9的缓冲溶液中的稳定性,从而比较其水解速率。其中,缓冲溶液由10mmol/l的磷酸二氢钠或磷酸氢二钠配置,再用氢氧化钠和盐酸调至上述指定的ph值。在6种氯苯乙腈内分别加入缓冲溶液后的溶液在23.0±0.5℃下避光保存120h,在0h、12h、24h、36h、48h、72h、96h和120h分别测定6种氯苯乙腈的剩余含量并进行对比。结果表明,6种氯苯乙腈在ph=7的条件下稳定性最好。
(氯化终止剂的确定)
实际上,氯化终止剂选自抗坏血酸、硫代硫酸钠、氯化铵、亚砷酸钠和亚硫酸钠中的一种以上。
最佳氯化终止剂的确定过程为:配置6个相同浓度(0.6μmol/l)的氯苯乙腈混合标准样品的水溶液,其中一个水样不加入氯化终止剂,其余溶液分别加入200μmol/l的抗坏血酸、硫代硫酸钠、氯化铵、亚砷酸钠(即偏亚砷酸钠)和亚硫酸钠,调节所有溶液的ph=7,在23.0±0.5℃下避光反应24h,再进行液液萃取,随后进入gc/ms分析。可得到峰面积最大时所用的氯化终止剂为亚硫酸钠。
加入氯化终止剂的目的为:通常消毒后的饮用水中含有0.05-4.0mg/l的余氯,余氯具有较强的氧化性,可能会与氯苯乙腈发生化学反应,从而影响检测结果的准确性,因此,应该选择合适的氯化终止剂终止氯化反应的进程,后继尽可能减少样品运输或存储过程中氯苯乙腈浓度的损失。
(萃取剂)
萃取剂为甲基叔丁基醚。
[分析条件优化]
实际上,在步骤(2)中,分析条件优化包括柱温箱升温程序、柱头压、进样量、进样口温度和色谱识别时间带的确定。
(柱温箱升温程序的确定)
气相色谱仪中柱温箱升温程序的确定过程为:对比6种氯苯乙腈的出峰时间、峰高和峰面积,在保证较短的总时间下,增大相邻峰的出峰时间间隔,尽可能提高峰高和峰面积,进而确定最佳的初始温度、终点温度、升温速度和各升温阶段末的保持时间。其中,初始温度通常为20-50℃,以保证样品中所有低沸点组分得到良好分离;终点温度应保证样品中的最后出峰组分的峰形尖锐且完全分离,同时不超过气相色谱仪的最高使用温度;升温速度应使得分析中各组分峰达到良好分离,由于6种氯苯乙腈的沸点和极性等性质各不相同,有的性质比较接近,以致难以分离,从而设置多个不同的升温速度的目的在于将6种氯苯乙腈更好地分开。
因此,本发明的柱温箱升温程序的确定方法可以克服6种氯苯乙腈的峰拖尾现象,使得各个氯苯乙腈实现完全分离,从而保证6种氯苯乙腈分别正常出峰。
(柱头压的确定)
气相色谱仪的柱头压为134.2±0.3kpa,优选为134.2kpa。
具体地:柱头压的确定过程为:在气相色谱仪其他参数不变的情况下,改变气相色谱仪参数中的柱头压,得到保证6种氯苯乙腈正常出峰时的柱头压即为最佳柱头压。
(进样量的确定)
气相色谱仪的进样量为1.0-10μl,优选为1.0-5.0μl,进一步优选为1.0μl。
具体地:最佳进样量的确定过程为:在气相色谱仪其他参数不变的情况下,将气相色谱仪设定为全扫模式,分别将进样量设定为1μl、2μl、3μl、4μl和5μl,单位进样量对应的6种氯苯乙腈总离子峰面积最大时的进样量为最佳进样量。
(进样口温度的确定)
气相色谱仪的进样口温度为100-250.0℃,优选为110-250.0℃,进一步优选为230.0℃。
具体地:进样口温度的确定过程为:进样口温度应使注入进样口的样品迅速汽化,在气相色谱仪其他参数不变的情况下,分别将进样口温度设定为100℃、130℃、150℃、170℃、200℃、230℃和240℃等,当6种氯苯乙腈的出峰达到良好分离时,峰面积最高时的进样口温度即为最佳进样口温度。
(色谱识别时间带的确定)
气相色谱仪的色谱识别时间带为0.1-1.0min,优选为0.2min。
具体地:色谱识别时间带的确定过程为:通常测定水样的过程中,由于样品基质的影响,目标物质的出峰时间与标准物质的出峰时间存在一定差异,其值在标准物质出峰时间前后偏移。由于6种氯苯乙腈中存在同分异构体,出峰时间比较接近,若时间带设置过宽,将不能实现各组分色谱峰的良好分离,若时间带设置过窄,则对样品基质的敏感性过高,无法正常测定水样中的6种氯苯乙腈。因此,在气相色谱仪其他参数不变的情况下,分别将时间带设定为0.1min、0.2min、0.4min、0.6min、0.8min和1.0min,当6种氯苯乙腈出峰达到良好分离时正常出峰的时间带,即为最佳色谱识别时间带。
因此,气相色谱仪的运行参数为:毛细管柱:型号为:rtx-5ms,柱长为:30m,内径为:0.25mm,膜厚度为:0.25μm;柱头压为:134.2±0.3kpa;进样量为:1.0-10μl;进样方式为:不分流进样;进样口温度为:100-250.0℃;进样时间为:1.0min;载气为:高纯氦气;载气流量控制方式为:压力控制;总流量为:31.5ml/min;柱流量为:2.31ml/min;线速度为:3312cm/min;吹扫流量为:6.0ml/min;数据采集、分析:gc/mssolution软件工作站;柱温箱升温程序:初始温度为:20-50℃,保持5.0min,再以25.00℃/min的速度升温至110℃,保持2.0min;再以35.00℃/min的速度升温至155℃,保持3.0min;再以20.00℃/min的速度升温至220℃,保持3.0min;色谱识别时间带为:0.1-1.0min。
质谱仪运行参数:离子源温度为:200℃,接口温度为:260℃;溶剂延迟时间为:3.0min;离子源为:电子轰击离子源(ei);电子能量为:70ev;检测模式为:选择离子检测(sim);扫描参数为:开始时间为:5.00min,结束时间为:19.00min,通道1:116.00m/z、通道2:151.00m/z、通道3:89.00m/z;开始时间为:12.12min,结束时间为:19.00min,通道1:150.00m/z、通道2:185.00m/z、通道3:152.00m/z、通道4:114.00m/z、通道5:73.00m/z和通道6:147.00m/z。
[运行测定]
运行测定包括标准曲线、方法检测限和回收率的确定。
(标准曲线的确定)
在测定6种氯苯乙腈的出峰时间之前,先建立6种氯苯乙腈的标准曲线,其具体过程为:
(a)、配置混合标准溶液:分别称取20mg的4-氯苯乙腈、20mg的2,3-二氯苯乙腈、20mg的2,4-二氯苯乙腈、20mg的2,5-二氯苯乙腈、20mg的2,6-二氯苯乙腈和20mg的3,4-二氯苯乙腈,溶于20ml甲基叔丁基醚中,混合配置成各个氯苯乙腈浓度相同(初始浓度均为1mg/ml)的混合标准溶液,并置于棕色瓶中;
(b)、用超纯水(millipore,电阻率为18mω·cm)将步骤(a)中的混合标准溶液配置成梯度标准校正液,梯度质量浓度分别为1μg/l、5μg/l、10μg/l、20μg/l、50μg/l、100μg/l和200μg/l;
(c)、采用气相色谱/质谱联用仪对上述梯度标准校正液进行批处理分析,利用labsolution工作站分别生成6种氯苯乙腈的标准曲线,6种氯苯乙腈的标准曲线图如图2至图7所示,其中,横坐标为氯苯乙腈的质量浓度(μg/l),纵坐标为氯苯乙腈的峰面积。信号强度在保留时间上的积分为峰面积,峰面积和氯苯乙腈的质量浓度成正比,由标准曲线可以得到标准的峰面积和质量浓度的关系,即在测未知样品时,用已知的峰面积可以得到未知样品中氯苯乙腈的质量浓度。
采用外标法建立的6种氯苯乙腈的标准曲线图,通过gc/ms对水样进行检测,如图8所示,横坐标为时间(min),纵坐标为离子碎片强度,无单位,4-氯苯乙腈的出峰时间可以为11.110±0.005min,优选为11.110min;2,4-二氯苯乙腈的出峰时间可以为11.680±0.005min,优选为11.680min;2,5-二氯苯乙腈的出峰时间可以为13.150±0.005min,优选为13.150min;2,6-二氯苯乙腈的出峰时间可以为13.375±0.005min,优选为13.375min;2,3-二氯苯乙腈的出峰时间可以为13.600±0.005min,优选为13.600min;3,4-二氯苯乙腈的出峰时间可以为14.385±0.005min,优选为14.385min。
6种氯苯乙腈在gc中分离后,ms检测到各种氯苯乙腈相应离子碎片的时间,因此,其出峰时间和仪器参数、物质沸点和极性等性质相关。
(方法检测限和回收率的确定)
采用本发明的检测方法,使得6种氯苯乙腈在分析过程中可实现良好分离,在1-200μg/l范围内线性良好(r^2>0.99),6种氯苯乙腈的回收率在87-95%之间;方法检测限(limitofmethoddetection,mdl)在100ng/l以下;相对标准偏差(relativestandarddeviation,rsd)小于10.0%。
其中,回收率是判断该检测方法是否可靠的判别标准,回收率越高,说明了检测结果值与理论值越接近,该检测方法越可靠;较高的方法检测限是指能够检测到较低浓度的氯苯乙腈;而较小的相对标准偏差是指多次检测结果相差不大,从而保证了检测结果的真实性。
以下结合实施例对本发明作进一步的说明。
实施例:
本实施例的同时检测饮用水中含氮芳香性消毒副产物-6种氯苯乙腈的方法包括如下步骤:
(1)、将饮用水的水样通过液液萃取预处理的方式进行富集。
具体地,取10ml饮用水的水样置于25ml玻璃小瓶中,加入200μmol/l的亚硫酸钠(作为氯化终止剂)脱氯后,调节该水样的ph=7,加入2.0g无水硫酸钠,手动振荡使其充分溶解;加入2.0ml甲基叔丁基醚(作为萃取剂),使用ika振荡器在2500r/min下振荡5.0min,静置10.0min,取1ml上层甲基叔丁基醚溶液置于进样小瓶内,进行gc/ms测定。
(2)、确定gc/ms的运行参数。
具体地,气相色谱仪的运行参数为:毛细管柱:型号为:rtx-5ms,柱长为:30m,内径为:0.25mm,膜厚为:0.25μm;柱头压为:134.2kpa;进样量为:1.0μl;进样方式为:不分流进样;进样口温度为:230.0℃;进样时间为:1.0min;载气为:高纯氦气;载气流量控制方式为:压力控制;总流量为:31.5ml/min;柱流量为:2.31ml/min;线速度为:3312cm/min;吹扫流量为:6.0ml/min;数据采集、分析:gc/mssolution软件工作站;柱温箱升温程序:初始温度为:50℃,保持5.0min,再以25.00℃/min的速度升温至110℃,保持2.0min;再以35.00℃/min的速度升温至155℃,保持3.0min;再以20.00℃/min的速度升温至220℃,保持3.0min;色谱识别时间带为:0.2min。
质谱仪运行参数:离子源温度为:200℃,接口温度为:260℃;溶剂延迟时间为:3.0min;离子源为:电子轰击离子源(ei);电子能量为:70ev;检测模式为:选择离子检测(sim);扫描参数为:开始时间为:5.00min,结束时间为:19.00min,通道1:116.00m/z、通道2:151.00m/z、通道3:89.00m/z;开始时间为:12.12min,结束时间:19.00min,通道1:150.00m/z、通道2:185.00m/z、通道3:152.00m/z、通道4:114.00m/z、通道5:73.00m/z和通道6:147.00m/z。
(3)、建立6种氯苯乙腈的标准曲线。
具体地,(a)、配置混合标准溶液:分别称取20mg的4-氯苯乙腈、20mg的2,3-二氯苯乙腈、20mg的2,4-二氯苯乙腈、20mg的2,5-二氯苯乙腈、20mg的2,6-二氯苯乙腈和20mg的3,4-二氯苯乙腈,溶于20ml甲基叔丁基醚中,混合配置成各个氯苯乙腈浓度相同(初始浓度均为1mg/ml)的混合标准溶液,并置于棕色瓶中;
(b)、用超纯水(millipore,电阻率为18mω·cm)将步骤(a)中的混合标准溶液配置成梯度标准校正液,梯度质量浓度分别为1μg/l、5μg/l、10μg/l、20μg/l、50μg/l、100μg/l和200μg/l;
(c)、采用气相色谱/质谱联用仪对上述梯度标准校正液进行批处理分析,利用labsolution工作站分别生成6种氯苯乙腈的标准曲线,其中横坐标为氯苯乙腈的质量浓度,纵坐标为氯苯乙腈的峰面积。
(4)、采用气相色谱/质谱联用仪分析水样,根据得到的色谱图和质谱图进行定性分析,然后采用外标法建立的6种氯苯乙腈的标准曲线确定水样中6种氯苯乙腈的出峰时间。
具体地,4-氯苯乙腈的出峰时间为11.110min,2,4-二氯苯乙腈的出峰时间为11.680min,2,5-二氯苯乙腈的出峰时间为13.150min,2,6-二氯苯乙腈的出峰时间为13.375min,2,3-二氯苯乙腈的出峰时间为13.600min,3,4-二氯苯乙腈的出峰时间为14.385min。
采用本发明的检测方法,使得6种氯苯乙腈在分析过程中可实现良好分离,在1-200μg/l范围内线性良好(r^2>0.99),6种氯苯乙腈的回收率为90%;方法检测限(limitofmethoddetection,mdl)在100ng/l以下;相对标准偏差(relativestandarddeviation,rsd)小于10.0%。
其中,4-氯苯乙腈的出峰时间在11.110±0.005min之内、2,4-二氯苯乙腈的出峰时间在11.680±0.005min之内、2,5-二氯苯乙腈的出峰时间在13.150±0.005min之内、2,6-二氯苯乙腈的出峰时间在13.375±0.005min之内、2,3-二氯苯乙腈的出峰时间在13.600±0.005min之内、3,4-二氯苯乙腈的出峰时间在14.385±0.005min之内均是可以的。
气相色谱仪中,柱头压在134.2±0.3kpa之内、进样量在1.0-10μl之内、进样口温度在100-250.0℃之内、色谱识别时间带在0.1-1.0min之内也是可以的。
上述对实施例的描述是为了便于该技术领域的普通技术人员能理解和使用本发明。熟悉本领域技术人员显然可以容易的对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中,而不必经过创造性的劳动。因此,本发明不限于上述实施例。本领域技术人员根据本发明的原理,不脱离本发明的范畴所做出的改进和修改都应该在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!