一种在氯漂白水溶液体系中准确检验酚类生成二噁英的方法与流程
本发明涉及一种氯漂白水溶液体系中准确检验酚类生成二噁英的方法,属于持久性污染物分析领域。具体来说是在天然酚类水溶液氯漂白体系中加入13c6标记的酚类用于漂白,得到漂白样品经提取净化后,利用高分辨气相色谱-高分辨质谱法对二噁英目标物进行定性定量分析,通过检测漂白生成的pcdd/fs,尤其是13c612c6-pcdd/fs,来判定酚类在此过程中是否能转化生成pcdd/fs,为pcdd/fs生成机理的研究提供直接依据。
背景技术:
二噁英(pcdd/fs)是斯德哥尔摩公约中首批持久性污染物之一,具有高毒性、环境持久性、长距离迁移性和生物累积性,根据其苯环碳上氢原子被氯原子取代的数目和位置的不同,pcdd/fs可总形成210种同类物,包括75种pcdds和135种pcdfs。pcdd/fs作为一种典型的非故意产生的持久性有机污染物,其污染控制难度较大,我国在制定履行《关于持久性有机污染物的斯德哥尔摩公约》国家实施计划过程中,根据不同排放源对pcdd/fs总排放量的贡献及发展趋势,将六大行业确定为我国pcdd/fs优先控制的重点行业,分别为废物焚烧行业、造纸行业(有氯漂白)、钢铁行业、再生有色金属行业、火化机和化工行业。
酚类是苯环上连接一个或多个羟基基团的化合物,由于酚类自身的氧芳环结构,可能成为pcdd/fs的前体来源物质。在造纸过程中,植物组织中含有种类丰富的酚类,经过化学制浆后降解生成大量相对游离的酚类,这些酚难以洗净而被带入漂白阶段,在含氯漂白剂的作用下,其中的酚类会转化为大量的可吸附性有机卤化物(aox),pcdd/fs属于aox的一种成分;酚类也是工业生产五氯酚、2,4-d、氯醌、三氯生等的起始原料,经过一系列氧化氯化的生产过程,在这些工业产品中也均能检测到pcdd/fs;另外在一些酚类的高温气相反应中运用在线gc-ms也观测到产物中存在pcdd/fs。
以上这些反应虽然条件各有不同,但在反应产物中均检测到了pcdd/fs的存在,可以间接证明酚类与pcdd/fs之间存在一定联系,在合适的条件下可以转化为pcdd/fs。但对于各个条件,尤其是纸浆氯漂白条件下酚类与pcdd/fs的机理研究并不成熟,究其原因,主要有:pcdd/fs作为一种超痕量持久性有机污染物,其可能广泛存在于环境中,而目前研究基本只分析其中的12c12-pcdd/fs天然化合物,并且文献中对于pcdd/fs的检测基本局限于17种2,3,7,8位取代的高氯代pcdd/fs,并不包括低氯代pcdd/fs和无对应提取内标的pcdd/fs同类物,其对于pcdd/fs的检测不论是种类还是同类物数量均不全面;而检测到的产物中的pcdd/fs也可能来自于:
(1)环境的本来存在的pcdd/fs污染,造成分析结果的假阳性;
(2)这些前体酚类物质中存在的微量低氯代pcdd/fs,由于它们转化为高氯代pcdd/fs的转化率比酚类、氯酚或其他前体物质高得多,所以即使含量少也极容易转化为高氯代pcdd/fs而被检测;
(3)不排除其他种类前体物质作为杂质存在于反应体系,或者对机理反应造成影响。
总的来说,目前在酚类生成pcdd/fs机理研究中,前体物质和反应产物中对所有种类pcdd/fs的检测不全面;而从机理本质出发,pcdd/fs的形成主要通过denovo和氯酚氯苯类等前体偶联形成,在一些工业高温过程中,这两种机理同时存在。酚类含有一个苯环,且存在羟基氧结构,而pcdd/fs是两个苯环并一个氧或两个氧的结构,在光激发、酶催化或纸浆氯漂白这种相对温和的条件(水溶液体系、存在大量酚类化合物、通常常温、有直接氯源)下,推断酚类若生成pcdd/fs可能的途径是通过氧化偶联而实现。为减少工业纸浆氯漂白过程中二噁英的生成和释放,研究二噁英生成的前体物质基础和动力学机制,是提出二噁英减排措施的核心依据,而检验酚类为生成pcdd/fs的前体物质是进行纸浆漂白条件下酚类与二噁英生成机理研究的先导基础。目前的研究还未有从酚类和pcdd/fs的分子结构入手,引入13c6标记的酚类和13c612c6-pcdd/fs,排除环境中天然pcdd/fs尤其低氯代pcdd/fs的干扰,直接确认酚类为生成pcdd/fs的前体物质的思路方法。
技术实现要素:
针对上述问题,本发明的目的在于提供在氯漂白水溶液体系中准确检验酚类生成二噁英的方法,其特征在于:包括以下步骤:
1.在所述氯漂白水溶液体系中使用天然酚类与13c6标记的混合酚类用于氯漂白,得到漂白样品;13c6标记的酚类的添加量为天然酚类用量的0.1~1%;
2.提取所得漂白样品中二噁英目标物,进行净化处理;利用基于高分辨气相色谱-高分辨质谱的方法对二噁英目标物进行定性定量分析。
步骤1中,所述的体系为18~35℃的超纯水溶液体系;所述的天然酚类为苯酚、愈创木酚或邻苯二酚中的一种或二种以上,天然酚类得用量为11.84~15.78mmoll-1。
所述氯漂白水溶液的氯漂白为次氯酸钠溶液漂白使有效氯用量为0.3~1.2g,ph值0~2,漂白时间5~180min,漂白完毕后加入10~25ml0.1~0.2moll-1的na2s2o3溶液用于淬灭残余有效氯,得到漂白样品。
步骤2中,所述的提取二噁英目标物,对漂白样品加入二噁英提取内标后,按漂白液体积的10~12%加入二氯甲烷液液萃取,共振荡萃取3次;对部分漂白样品中少量的固形物,用15~20ml二氯甲烷超声3次,合并萃取液和超声液,用无水硫酸钠除水,旋蒸浓缩至2~3ml,进行净化处理,随后浓缩加进样内标溶液定容。
步骤2中,所述的高分辨气相色谱-高分辨质谱分析,在高分辨质谱选择离子监测模式相应通道内加入13c612c6-pcdd/fs的两个特征离子(lm1和lm2)进行监测,经过优选,dbf的lm1和lm2为174.0776和175.0776,mocdfs的lm1和lm2为208.0387和210.0357,dicdfs的lm1和lm2为241.9992和243.9962,tricdfs的lm1和lm2为275.9602和277.9572,tcdfs的lm1和lm2为311.9183和309.9212,pecdfs的lm1和lm2为345.8793和347.8763,hxcdfs的lm1和lm2为379.8403和381.8374,hpcdfs的lm1和lm2为413.8013和415.7984,ocdf的lm1和lm2为449.7594和447.7624;dbd的lm1和lm2为190.0726和191.0726,mocdds的lm1和lm2为224.0336和226.0306,dicdds的lm1和lm2为257.9941和259.9911,tricdds的lm1和lm2为291.9551和293.9521,tcdds的lm1和lm2为327.9132和325.9161,pecdds的lm1和lm2为361.8742和363.8712,hxcdds的lm1和lm2为395.8352和397.8323,hpcdds的lm1和lm2为429.7963和431.7933,ocdd的lm1和lm2为465.7543和463.7573。
步骤2中,所述的二噁英目标物的定性定量分析,对13c612c6-pcdd/fs定性采用保留时间(或相对保留时间)和两个特征监测离子的离子丰度比进行,定量采用同位素稀释内标法,其相对响应因子采用对应系列标准溶液的测定中12c12-pcdd/fs天然化合物和13c12-提取内标物之间的相对响应因子,从而计算生成13c612c6-pcdd/fs化合物的含量。
经过步骤1、步骤2后,得到了酚类经过此氯漂白过程后生成的pcdd/fs含量,从检测到13c612c6-pcdd/fs的存在来判定酚类在此氯漂白过程中生成了pcdd/fs。
本发明的优点和有益效果为:本方法在现有判定酚类作为pcdd/fs前体物质的机理实验研究方法不足的基础上,结合纸浆氯漂白和其他氯漂白体系,提出的一种简易准确的判定方法。
(1)在实验设计上,该方法从pcdd/fs和酚类的结构本质出发,创造性地加入13c6标记的酚类作为前体物质,加入代表性的13c6标记的苯酚,同时考虑到其高成本,仅加入少量的13c6标记的苯酚将之与天然酚类混合漂白,节约实验研究成本的同时又避开了大量天然化合物的干扰,将酚类生成pcdd/fs的本质特征清晰明了地反映出来。
(2)这种13c6标记的酚类同位素示踪的方法对于研究其他工业过程中由酚类生成pcdd/fs的生成路径和机制具有启示作用。
(3)在分析方法上,对氯漂白反应生成的所有pcdd/fs种类均通过高分辨气相色谱-高分辨质谱进行分析,减少了干扰和假阳性结果,定性准确;定量上,由于添加的13c6标记的酚类含量相对极少,考虑pcdd/fs为一种超痕量有机污染物,在常温水溶液体系中由酚类转化为pcdd/fs的转化率是比较低的,因此由添加的13c6标记的酚类相互之间生成的13c12-pcdd/fs是极其微量的,因此其对提取时添加的13c12-pcdd/fs提取内标的影响可以忽略不计,因此,采用本方法中的同位素稀释内标法可以准确定量生成的pcdd/fs,尤其对关键产物13c612c6-pcdd/fs可以实现定性定量,这种高分辨方法分离分析效率高、选择性好、检出限低,可为机理研究提供直接证据。
(4)另外,加入严格的质控和对照实验,对反应中由天然酚类相互之间生成的12c12-pcdd/fs天然化合物的同步定性定量,结合13c612c6-pcdd/fs的结果,可为此检验方法提供进一步佐证。
附图说明
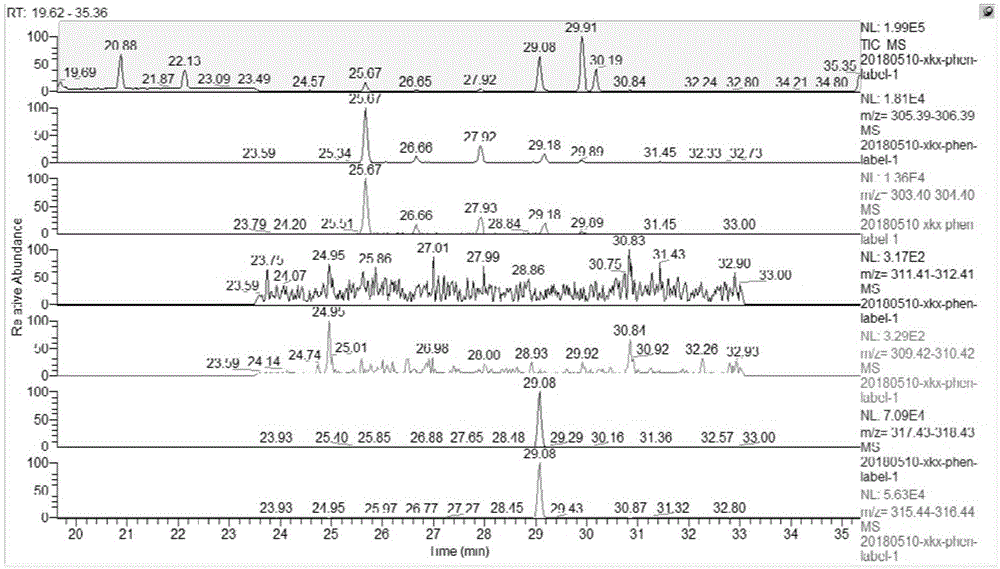
图1为苯酚混合漂白生成pcdd/fs的tcdf选择离子流图。
具体实施方式
本发明的目的在于提供一种水溶液体系中准确检验酚类生成二噁英的方法,以下实施例用于对本发明进行进一步说明,应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明的范围。
本发明提供的水溶液体系中准确检验酚类生成二噁英的方法,包括以下步骤:
1.天然酚类与13c6标记的酚类混合用于氯漂白:将0.5g酚类和0.5mg13c6-苯酚置于三口烧瓶中,加入超纯水325ml,用浓hcl调节体系ph值至1,加入次氯酸钠溶液漂白剂使有效氯用量为0.5g,25℃水浴高速搅拌反应60min,反应结束后加入15ml0.2moll-1na2s2o3溶液终止反应。
2.反应产物提取与净化:对漂白液体,在其中加入10μl壬烷配制的浓度为100pg/ul的13c12标记的dbf,2-mocdf,2,3-dicdf,2,3,8-tricdf,dbd,2-mocdd,2,3-dicdd,2,3,7-tricdd提取内标溶液ⅰ,和10μl壬烷配制的13c12标记的2,3,7,8-tcdf,1,2,3,7,8-pecdf,2,3,4,7,8-pecdf,1,2,3,4,7,8-hxcdf,1,2,3,6,7,8-hxcdf,1,2,3,7,8,9-hxcdf,2,3,4,6,7,8-hxcdf,1,2,3,4,6,7,8-hpcdf,1,2,3,4,7,8,9-hpcdf,2,3,7,8-tcdd,1,2,3,7,8-pecdd,1,2,3,4,7,8-hxcdd,1,2,3,6,7,8-hxcdd,1,2,3,4,6,7,8-hpcdd,ocdd提取内标溶液ii,除13c12标记的ocdd浓度为200pg/ul,其余14种13c12标记的pcdd/fs浓度为100pg/ul;按漂白液体积的10%加入二氯甲烷液液萃取,共萃取3次;对漂白产生的固形物,用15ml二氯甲烷超声3次,合并萃取液和超声液,用无水硫酸钠除水,旋蒸浓缩至2ml,进行硅胶柱和氧化铝柱净化处理,净化液旋蒸浓缩至1~2ml,n2吹至近干,加入10μl浓度为100pg/ul的13c12标记的1,2,3,4-tcdd和1,2,3,7,8,9-hxcdd进样内标壬烷溶液,涡旋混匀,待分析。
3.高分辨气相色谱-高分辨质谱分析:色谱柱为rtx-5ms柱(60m×0.25mmi.d.×0.25μmdf);载气为氦气(纯度≥99.999%),恒流模式,流速1.0ml/min;不分流进样,进样体积1μl,进样口温度280℃;升温程序为初始温度120℃,保持1min,以43℃min-1上升到220℃,保持15min,然后以2.3℃min-1上升到250℃,以0.9℃min-1上升到260℃,最后以20℃min-1上升到310℃,保持20min;传输线温度280℃。高分辨质谱为ei电离源,离子源温度260℃,电离能37ev,离子化电流600μa,离子加速电压8000v,分辨率10000,以选择离子监测(sim)模式选择12c12-,13c612c6-,13c12-全系列二噁英的两个特征离子进行监测。经过优选,通道内pcdd/fs各特征离子质量数汇总如表1所示。
表1
4.目标物定性方法:采用保留时间(或相对保留时间)和两个特征监测离子的离子丰度比进行定性。对于有对应提取内标参考物的二噁英同类物,其保留时间与标准溶液中相应同类物保留时间偏差在±0.3%之内。对于无标准参照物的二噁英同类物,不能精确定性二噁英芳环上氯取代的位置,但可以在对应氯取代通道的时间窗口范围内对各氯代二噁英色谱峰进行识别,确定其为几氯代的二噁英;所有二噁英同类物的两个监测离子在此保留时间窗口内同时存在,各氯代pcdd/fs离子丰度比与理论离子丰度比一致,相对偏差在±15%之内。同时满足上述条件的色谱峰定性为二噁英同类物。其中样品中13c612c6-pcdd/fs同类物的定性参照相应12c12-pcdd/fs和13c12-pcdd/fs同类物。
5.目标物定量方法:通过系列标准溶液的测定分析,根据其中12c12-pcdd/fs天然化合物、13c12-提取内标物和13c12-进样内标物三者浓度和相应质谱响应峰面积,计算12c12-pcdd/fs天然化合物和13c12-提取内标物之间的相对响应因子(rrf),以及13c12-提取内标物和13c12-进样内标物之间的响应因子(rf),具体方法如下:
式中:rrf——12c12-pcdd/fs天然化合物相对于13c12-提取内标的相对响应因子;
a1n——校正标准溶液中12c12-pcdd/fs天然化合物的第一个特征质量数m1离子的峰面积;
a2n——校正标准溶液中12c12-pcdd/fs天然化合物的第二个特征质量数m2离子的峰面积;
cl——校正标准溶液中13c12-提取内标的浓度,pg/ul;
a1l——校正标准溶液中13c12-提取内标的第一个特征质量数llm1离子的峰面积;
a21——校正标准溶液中13c12-提取内标的第二个特征质量数llm2离子的峰面积;
cn——校正标准溶液中12c12-pcdd/fs天然化合物的浓度,pg/ul。
式中:rf——13c12-提取内标相对于13c12-进样内标的响应因子;
a1l——校正标准溶液中13c12-提取内标的第一个特征质量数llm1离子的峰面积;
a2l——校正标准溶液中13c12-提取内标的第二个特征质量数llm2离子的峰面积;
cis——校正标准溶液中13c12-进样内标的浓度,pg/ul;
a1is——校正标准溶液中13c12-进样内标的第一个特征质量数llm1离子的峰面积;
a2is——校正标准溶液中13c12-进样内标的第二个特征质量数llm2离子的峰面积;
cl——校正标准中13c12-提取内标的浓度,pg/ul。
在样品提取净化前,定量添加13c12-提取内标,进样分析前加入一定量13c12-进样内标,根据在系列校正标准溶液中测定的相对响应因子(rrf)和响应因子(rf),计算样品中12c12-pcdd/fs天然化合物的含量(qnat)。由于13c612c6-pcdd/fs与12c12-pcdd/fs天然化合物结构与性质的相似性,对于反应过程中生成13c612c6-pcdd/fs含量(qiso)的计算采用系列标准溶液的测定分析中12c12-pcdd/fs天然化合物和13c12-提取内标物之间的相对响应因子(rrf)用于计算。
式中:qnat——分析样品中12c12-pcdd/fs天然化合物的量,pg;
a1n——12c12-pcdd/fs天然化合物的第一个特征质量数m1离子的峰面积;
a2n——12c12-pcdd/fs天然化合物的第二个特征质量数m2离子的峰面积;
ql——13c12-提取内标的添加量,pg;
a1l——13c12-提取内标的第一个特征质量数llm1离子的峰面积;
a21——13c12-提取内标的第二个特征质量数llm2离子的峰面积;
rrf——12c12-pcdd/fs天然化合物对于13c12-提取内标的相对响应因子。
式中:qiso——分析样品中13c612c6-pcdd/fs的量,pg;
a1n——13c612c6-pcdd/fs的第一个特征质量数lm1离子的峰面积;
a2n——13c612c6-pcdd/fs的第二个特征质量数lm2离子的峰面积;
ql——13c12-提取内标的添加量,pg;
a1l——13c12-提取内标的第一个特征质量数llm1离子的峰面积;
a21——13c12-提取内标的第二个特征质量数llm2离子的峰面积;
rrf——12c12-pcdd/fs天然化合物对于13c12-提取内标的相对响应因子。
对于样品前处理过程中13c12-提取内标的回收率(r)的计算,具体方法如下:
式中:r——13c12-提取内标的回收率,%;
a1l——13c12-提取内标的第一个特征质量数llm1离子的峰面积;
a2l——13c12-提取内标的第二个特征质量数llm2离子的峰面积;
qis——13c12-进样内标的添加量,pg;
a1is——13c12-进样内标的第一个特征质量数llm1离子的峰面积;
a2is——13c12-进样内标的第二个特征质量数llm2离子的峰面积;
rf——13c12-提取内标相对于13c12-进样内标的响应因子;
ql——13c12-提取内标的添加量,pg。
实施例1
将0.5g苯酚与0.5mg13c6-苯酚按照上述步骤进行漂白,提取,净化和分析,检测计算样品中的12c12-pcdd/fs天然化合物和13c612c6-pcdd/fs。同时进行仅加0.5g苯酚的漂白实验和苯酚未漂白的空白实验作为对照组,所有反应条件、前处理和仪器分析步骤均相同。均平行2份。内标回收率25~73%,仪器检出限和方法检出限如表2所示。
表2
检测发现,苯酚空白含有dbf2368.85pg/g,这是平常方法仅检测高氯代二噁英所不能检测的。而两组漂白样品生成的12c12-pcdd/fs含量基本相当,总pcdd/fs含量是空白苯酚pcdd/fs含量的2.98倍。由于存在反应系统和分析过程的误差,因此无法确定所生成的二噁英是来自于苯酚本身还是其中所含有的dbf杂质,因此需要加入同位素的实验进行进一步佐证,从而为后续机理实验和最终减排措施的提出奠定基础。
仅加苯酚漂白的样品在13c612c6-pcdd/fs通道内不存在对应的pcdd/fs峰,符合实验预期。而苯酚混合漂白的样品在13c612c6-pcdd/fs通道内基本不存在对应的pcdd/fs峰,如图1所示的苯酚混合漂白生成pcdd/fs的tcdf选择离子流图,在12c12-pcdd/fs和13c12-pcdd/fs通道内均存在明显的色谱峰,但是13c612c6-pcdd/fs通道内却不存在明显的色谱峰,证明未有明显13c612c6-pcdd/fs生成;在13c612c6-pcdd/fs其他氯代通道内,少数对应保留时间窗口内存在色谱峰,但是积分结果显示其离子比例不符合范围,因此其非有效pcdd/fs峰;也有部分色谱峰对应无相应提取内标,其离子比例符合范围,但色谱峰接近基线,且对应的天然pcdd/fs通道内无此峰存在,或另一平行样品中并无此峰,故其也非有效的pcdd/fs峰。因此综合来看,在此苯酚混合漂白的样品中未检测到13c612c6-pcdd/fs。
由此看出,如果单用天然的酚类进行实验,以检测反应生成的天然pcdd/fs而判定酚类是pcdd/fs的前体物质,即使考虑了酚类中固有的杂质,但由于存在转化率、环境污染等因素的影响,其证据和结论也是不够充分的。而若用本发明的方法,根据实际条件可以对配比和反应条件进行改变,以13c612c6-pcdd/fs的生成作为判定依据(本实验条件可能投入13c6-苯酚含量和比例较小,或存在仪器检出限还相对过高而使未检出13c612c6-pcdd/fs。但如若此种情况存在,也可看出酚类直接转化为二噁英的效应是非常低的,甚至是可以忽略的;在氯漂白条件下,酚类对二噁英生成的影响可能来自于酚类芳环自身会消耗一部分氯,从而影响二噁英的生成。此处的实验条件和结果并不影响此法应用于其他工业过程中关于酚类和二噁英生成机理的研究,尤其涉及热处理的焚烧和烧结等),与直接用天然酚类用于实验相比,有效地避免了杂质和环境的污染;与全部用13c6-苯酚用于实验相比,不仅节约了实验成本还有效地避免了实验环境中用于分析时加入的13c12-pcdd/fs提取内标污染系统,对检测13c12-pcdd/fs存在的影响。
- 还没有人留言评论。精彩留言会获得点赞!