一种染料中多氯苯酚的检测方法与流程
本发明属于分析化学
技术领域:
,尤其涉及染料中多氯苯酚的检测方法。
背景技术:
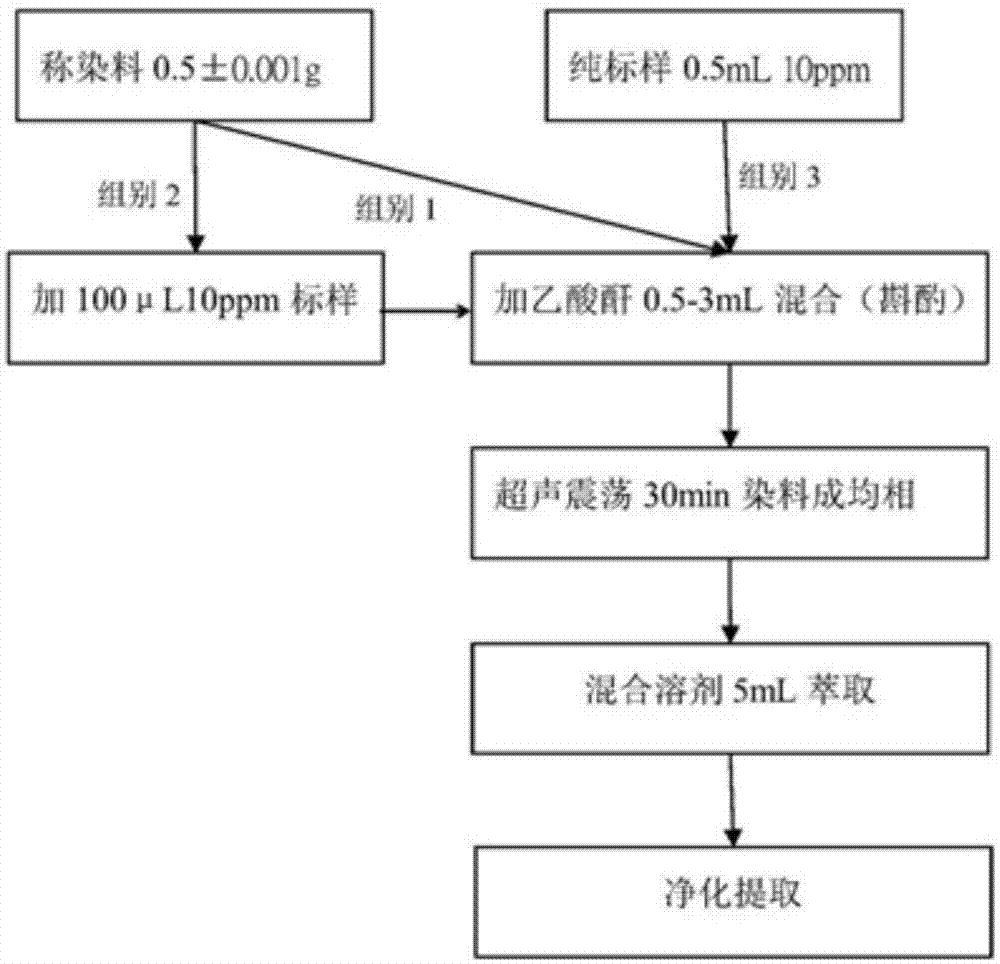
:五氯苯酚(简称:pcp),外观为白色薄片或结晶状固体,常含一分子结晶水,稍热有极强辛辣臭味,五氯苯酚是纺织品、皮革、木材等产品中常见的防腐剂,燃烧时会转变为二恶英类剧毒物质。经动物实验证实,pcp为生物致癌性的剧毒物质,并会累积于生物体内,造成不可逆的影响。四氯苯酚在杀虫剂上使用较为广泛,在国际癌症研究机构(iarc)中也有提出其对生物的影响,虽未确实对人体的影响,但也不容忽视。而在2016年,全球性绿色环保组织oekotexstandard100于有害物的控管条例中也新增了以往被忽视的一氯苯酚与二氯苯酚的含量限制。以往环保规范限制大多局限于成品中,因此目前的检测方法也仅止于纺织品(xp-g08015)与皮革(iso17070)的标准检测法。并且,关于皮革和纺织品中一氯苯酚、二氯苯酚等9种同分异构体和四氯苯酚与五氯苯酚的同时测定还未见发布相关标准方法。近年来环保议题逐渐被关注,不仅止于成品,其原物料的有害物成分亦开始着重,中华人民共和国于2009年颁布的检测国家标准也仅限制于染料中四氯苯酚与五氯苯酚的检测(gb/t24166-2009)。目前常用的检测方法有:iso17070:2006《皮革中五氯苯酚含量检测方法》;国家标准gb/t18414.2-2006《纺织品含氯苯酚的测定》;国家标准gb/t24166-2009《染料中氯苯酚的测定》;然而上述检测方法,存在以下不足之处:①目前技术对多氯苯酚色谱图积分连峰造成判图定量误判;②以现有方法萃取染料杂质难以去除;③现有染料萃取方法稳定度不高并且溶剂对染料溶解问题不够完善。技术实现要素:本发明针对现有技术的不足,本发明提供了一种萃取溶液量少,废液量低的染料中多氯苯酚的检测方法,该检测方法可以克服二氯苯酚、三氯苯酚色谱图积分连峰造成判图定量误判的困扰。为实现以上的技术目的,本发明将采取以下的技术方案:一种染料中多氯苯酚的检测方法,包括以下步骤:首先,将待测染料样品经过与乙酸酐混合反应后得到液相;然后加入混合溶剂形成均相溶液后,添加样品内标物;接着通过加入硫酸钠水溶液进行萃取分层,获得的萃取上层液经过离心、过滤分离后通过气相质谱仪检测氯苯酚含量。进一步的,染料中多氯苯酚的检测方法具体步骤如下:(1)乙酰化:将待测染料样品与乙酸酐进行混合,并在超声振荡作用下,使待测染料样品中还原基团(氯苯酚)与乙酸酐充分反应并溶解,不参与反应的染料剩余物位于液相底层;混合好的样品经过超声振荡30min以上;(2)提取:抽取步骤(1)得到的液相,并加入混合溶剂充分混合,然后加入样品内标物形成均相的样品溶液;所述样品内标物的结构与多氯苯酚相近,且在检测温度范围内能出现,并且不与待测样品、淬取溶剂反应影响到萃取过程的损失与干扰;要求待测染料样品与混合溶剂形成均相,以确保染料与溶剂之间充分溶解(根据不同染料结构、分散剂的种类对混和时间、加入溶解液做调整)。其中内标物在溶剂的含量为10±0.05ppm,纯度高于99.9wt.%。(3)净化:在步骤(2)得到的样品溶液中,加入硫酸钠水溶液形成萃取分层,萃取样品溶液中残留的染料;透过硫酸钠水溶液及无水硫酸钠做净化处理,降低干扰分析物的讯号强度。(4)分离:抽取萃取分层中上层的有机相样品,先经过离心机离心分离去除杂质,接着经过滤网过滤得到澄清液;(5)检测:将步骤(4)得到的澄清液通过气相质谱仪进行检测。作为优选,所述混合溶剂为正己烷、甲苯、异丙醇、丙醇中至少两种的混合物。作为优选,所述样品内标物为四氯化邻甲氧基苯酚。作为优选,步骤(3)中萃取净化的温度为200~250℃。根据以上的技术方案,相对于现有技术,本发明具有以下的优点:本发明针对于染料中多氯苯酚检测与其衍生物,其样品回收率达90-110%,并且重复检测其相对标准偏差(relativestandarddeviation,rsd)≤3.0%(样品数,n=3)。附图说明图1为本发明所述染料中多氯苯酚的检测方法的流程示意图;图2为应用本发明染料中多氯苯酚的检测方法的气相色谱-质谱仪tic图。具体实施方式附图非限制性地公开了本发明所涉及优选实施例的结构示意图;以下将结合附图详细地说明本发明的技术方案。说明:每一个样品必须添加待测成分(内标法或外标法)以决定该样品的回收率及检测极限;对样品的待测物出现背景值较多干扰的情况,并造成内标物回收率的检测限低于0.05ppm的样品,需要基质净化过程,但大多数环境及废弃物样品的萃取法,在分析前均需先行处理;当样品干扰值高于被测物的情况或导致保留时间偏移时,应加入外标物以确认出锋位置。如检测讯号超过检线的范围,则必须稀释待测物溶液并重新分析;每五种不同样品中须做重复检测其中几组数据以确保准确性以及样品的重现性。检测限(limitofdetection,lod)计算公式:d=3n/s(1)式中:n——噪音;s——检测器灵敏度;d——检测限;灵敏度的计算公式为:s=i/q(2)式中:s——检测器灵敏度;i——信号响应值;q——进样量;将式(1)和式(2)合并,得到下式:d=3n×q/i(3)式中:q——进样量;n——噪音;i——信号响应值;i/n即为该进样量下的信噪比(s/n),该信噪比可通过工作站对图谱进行自动分析获得,一般的色谱或质谱工作站都可进行信噪比分析计算。一、适用范围:本方法适用于分析染料基质中十九种氯苯酚成分。如需要较高的层析分辨率或者已参照本方法进行一次或多次净化步骤所得的萃取液时,建议使用分离管柱、过滤滤口或其他基本处理工艺进行净化。本方法的检测极限并非一成不变,其结果会随样品中基质的不同而有所差异。二、干扰:本方法的干扰来源归纳为三大类:①受污染的溶剂、试剂或样品处理用具;②受污染的气相层析使用的载流气体、零件、管柱表面或检测器表面;③环境湿度变化与温度变化。若遵循一般实验室操作规范,并且维持样品与操作环境管控,可大致避免第一类的干扰。然而染料测试当中,主要是样品基质所带来的干扰为最大,以及其他副产物的干扰物。某些复配染料较为复杂的样品需在本方法的基础上增加另外净化步骤处理样品,以达到确认及定量分析所要求的纯度。有机气农药(有机磷、有机硫等)在使用宽口毛细管柱分离时同时流出,也可能在萃取过程中同时萃取出来,成为本方法的干扰物。倘若当样品杂质过量而影响被检测物样品时,需增加净化步骤,将其余非待测物的有机气农药以及其余干扰物质去除。三、设备:1)玻璃器皿:参见超音波震荡萃取法、硅胶管柱净化与碳净化法的相关规格。2)气相质谱仪——气相层析分析系统必须兼具管端,分流-非分流式注射系统及其他相关附件,包括注射针、分析管柱、气体、电子捕捉侦测器、记录/积分器或数据处理系统。3)气相层析仪注射系统为重要部分,若受到污染、具化学活性或过高温的注射器均可能导致待测物裂解。4)气相层析仪的分析条件:①管柱-30m×0.25或0.32mm内径的熔硅毛细管柱,内覆se-54(柱子型号为db-5ms或相似管柱),膜厚度为0.25μm,窄口式管柱应装置于分流——非分流式注射系统。(安捷伦气象色谱柱,agilentj&wgccolums)管柱规格并非限制各实验室不得使用其他类似的管柱,实验室可使用其他类似之毛细管柱,只要有完整的书面数据证明其适用性。②离子源型号为:气象质谱仪gcms-qp2010(岛津,shimadzu);③进样器型号:自动进样器aoc-20its(岛津,shimadzu)5)气相质谱仪的分析条件①管柱条件:载流气体(氦气):5.74±0.5kpa;注射部温度:250±50℃;最终温度:280±50℃;最初温度:80±50℃;升温设定:80℃至280℃,以每分钟10±2℃升温。②离子源条件:离子源温度:230±50℃;接口温度:290±50℃;溶剂延迟:5±1分钟;电离能量:70±5ev。6)结果计算本申请公开的检测方法的检测极限为0.5ppm(mg/kg),检测样品的鉴定除其锋面积与浓度的计算,待测物的检测极限也需考虑,当讯号比(讯号/噪声)低于10时将不予计算。并且,若待测物的多氯苯酚出锋位置偏移时,须加入一外标样再做一次检测以确保萃取的准确性。检测值表示至小数点后两位,公式换算如下:χ——样品的待测含量,毫克每千克(mg/kg);a1——标准样品中多氯苯酚乙酸酯的峰面积(或峰高);v——样品液体积,毫升(ml);c——标准样品中多氯苯酚的含量,毫克每千克(mg/kg);a——样品液体中多氯苯酚乙酸酯的峰面积(或峰高);m——最终样品液试样量,克(g)。四、试剂与药粉1)所有测试分析过程均必须使用试药级或残量级(99.9wt%以上)的化学试药,并且所有试药均符合美国化学会分析试剂委员会所订的规范,分析实验用水规格和试验方法(gb/t6682-1992,neqiso3696:1987)不含有机物的二级水。2)溶剂与试药:参阅连续液相-液相萃取法、超声波震荡萃取法:混合溶剂及乙酸酐。所有溶剂均应为残量级或相等级次的试剂,每一批次溶剂均应分析确定不含非被萃物,以防止干扰。3)净化过程使用的无水硫酸钠:使用前,以200℃以上净化至少1小时;4)储备标准溶液:①以天平秤取0.1000±0.0010g的已知参考物质,溶于标配溶剂中,并稀释100ml的容量瓶中,以制备1000±0.001mg/l的储备标准溶液。如参考物质的纯度>99wt.%时,其重量可不校正而直接计算储备标准溶液的浓度。②将上述储备标准溶液移至有四氟乙烯内衬螺旋盖的瓶中并用封口膜封住接盖口,以-30℃冷藏避光贮存;此储备标准溶液必须经常检测是否有裂解或挥发情形发生,尤其是由此溶液来制备标准溶液,并每三个月或确认浓度有变时应实时更换。5)内标准品:使用本申请公开的方法时,分析人员必须选择一种或多种内标准品,其样品结构必须与被测物质性质相似,同时证明:该内标准品不受方法或基质干扰并不参与萃取过程反应。四氯化邻甲氧基苯酚(tetrachloroguajacol,cas号:2539-17-5)作为内标准品效果最佳。五、样品与溶剂保存方式:1)确认采样容器清洁后,再以溶剂润湿。样品容器材质需为玻璃等不干扰萃取物的材料。3)由于某些特定染料在受潮时、在萃取过程中不易分解,应将样品置于固定湿度环境储存以避免萃取困难及不必要的水份干扰。本发明创新点在于:以往人们关注的只有自身的安全并不在意环境的危害因此以往萃取方式与标准皆以成品作为主,然而环保意识对起始原物料的渐渐重视,而针对原物料(染料)的分析多氯苯酚萃取方法还没有一个明确的检验方式,本方法克服上述缺陷,实现了对染料更准确的分析。不管染料种类皆可通过此方法做调整而达到要求,较以往的纺织品检验更佳。在多次实验结果中发现,此萃取方法回收率高达九成以上,更进一步提供了针对染料准确萃取分析。实验过程相较以往的淬取时间较为快速,并且该淬取方法产生的废弃物较少,相对传统的方法更环保。面列举的具体实施例,是对本发明作进一步详细的说明,这些实施例仅用于说明本发明的目的,不以任何方式限制本发明所包含内容的范围。实施例选择试样描述本实验之萃取回收率代表试样选择以分散染料为主,主要系其对于溶剂的溶解最为困难并且其为市面上染料种类的最大宗。其他种类染料也经由此方法测试过,其萃取效果与分散染料并无太大差别,但其他染料部分杂质较少可视情况斟酌加入适当的溶解液做调整。试样描述本实验中以三种组合为实验考虑背景样品、背景样品加标样及标样,并且每组数据以重复三次操作。如是一般检测物在每次测量时应当再加一支溶剂样做为污染可能之考虑。如下:组别1.背景样品:1138红色(rubinexftb,2017/10/20出产,龙盛)(n=3):染料分子极其含量:组别2.外加标样浓度:5ppm,取100μl至背景样品(n=3);组别3.标样:5ppm0.5ml;试验日期:2018/03/09;1.试验流程1)染料0.5000±0.0001g至玻璃容器中,加入乙酸酐(过量)混合;2)将玻璃容器至于超音波震荡机至少30min;3)加入萃取液混合;4)加入硫酸钠水溶液(过量)与萃取液混合;5)加入1g无水硫酸钠混合;6)混合好后至离心机以至少1000rpm的速度离心分离至少5min;7)取上清液并用过滤头(0.22μm)过滤,即完成萃取步骤。2.试验结果1)本实验回收率系以标准曲线计算出的公式以加入标样的样品扣除空白组而得,为确保准确度每次重复三次以上,并在每一次过程中确认样品损失情况,以3倍信噪比计,本方法的检出限均为0.5mg/kg。2)每种氯苯酚回收率高达90-110%,且误差值(rsd)皆于3%以内。3)机台本身rsd值见表3。表1:多氯苯酚之组分资料:组分分子式分子量沸点(℃),常压一氯苯酚c6h5clo128.56175-176二氯苯酚c6h4cl2o163.00210三氯苯酚c6h3cl3o197.44246四氯苯酚c6h2cl4o231.89164/3.059kpa五氯苯酚c6hcl5o266.34309-310图2气相色谱图(tic)的多氯苯酚检测物1-19种组分(cp=一氯苯酚;dicp=二氯苯酚;tcp=三氯苯酚;tecp=四氯苯酚;pcp=五氯苯酚)。表2:多氯苯酚回收率与误差值表格表3:机台误差率表格标题保留时间(分钟,min)峰面积报告18.546699006报告28.545698735报告38.547700933平均值8.546699558相对标准偏差rsd(%)0.0120.1713.试验讨论(一)确认被测物的滞留时间或峰型加以辨识:定量方式则以该待测物特性尖峰下的面积与标曲线上换算浓度而得,并以标准品的出峰时间相互呼应互作比较而得。(二)滞留时间容许范围,以多次测定所得的滞留时间的标准偏差乘以5计算而得。(三)如被测物的样品出峰时间过于偏差需加入外标物以相同前处理手法以确认出锋位置的变动,当过于偏差出锋位置不予计算。(四)每次进样前先确认仪器是否为正常状态,并检查耗材使用状态以排除不必要的检测问题。以上所述的具体实施例,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1