一种HPLC测定单链取代二甲基苄基氯化铵含量的方法与流程
本发明涉及色谱分析领域,更具体地,涉及一种hplc测定单链取代二甲基苄基氯化铵含量的方法。
背景技术:
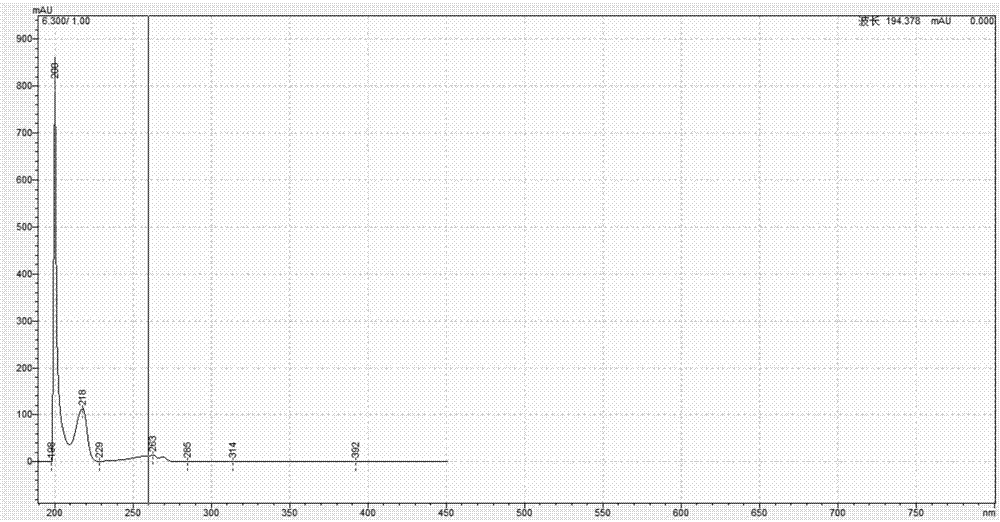
:季铵盐类化合物是一类重要的阳离子表面活性剂,具有一定的杀菌作用,因其杀菌谱广、作用强等优点被广泛应用于消毒产品、日化产品、印染、石油化工等领域。长链烷基季铵盐是季铵盐类化合物中重要的分支,其代表物质有苯扎氯铵(洁尔灭)、苯扎溴铵(新洁尔灭)、苄索氯铵等。其中,苯扎氯铵、苯扎溴铵在《中国药典》(2015)、usp30、bp2013、jpxvi各国药典均有记录,在临床领域应用十分广泛。上述这些物质结构均为二甲基苄基取代氯化铵,仅单链取代基不同,基本信息如表1所示:表1常见单链二甲基苄基氯化铵的基本信息单链烷基取代二甲基苄基氯化铵作为季铵盐类表面活性剂的主要品种,一直是人们关注的重点。对于其性能改进的研究工作主要体现在通过化学键联结的方法提高表面活性。近年来,c18-二甲基苄基氯化铵、苄索氯铵、椰油烷基二甲基苄基氯化铵等新型二甲基苄基氯化铵层出不穷。随着研究的不断深入和新型季铵盐化合物的应用日趋广泛,质量控制过程中对其含量测定的需求越来越多。目前针对季铵盐类化合物的含量测试主要有化学分析法和仪器分析法。化学分析法原理是利用季铵盐与沉淀剂或阴离子试剂的化学反应,适用于测定已知相对分子质量的单一季铵盐含量测定。化学分析法的优点是普适性强,缺点是操作复杂、干扰严重、准确度不高。仪器分析法最常见的有液相色谱和气相色谱法,分析原理是利用标准对照品拟合曲线,外标曲线法定量。仪器分析法的优点是高效、快速、准确度高,缺点是标准对照品难以获得,方法普适性不高。因此,为了更好的把控生产、销售和使用过程中的质量,需要建立一种简单、快捷、准确且普适性强的含量测试方法。技术实现要素:为解决现有技术的缺点和不足之处,本发明的目的在于提供一种hplc测定单链取代二甲基苄基氯化铵含量的方法。本发明的目的通过如下方案实现:一种hplc测定单链取代二甲基苄基氯化铵含量的方法,包括如下步骤:1)利用hplc建立c12-二甲基苄基氯化铵的标准工作曲线;2)在与步骤1)相同条件下用hplc检测含单链取代二甲基苄基氯化铵类化合物的试样;比对所述试样hplc图谱中各色谱峰的uv图谱和c12-二甲基苄基氯化铵的uv图谱以确认所述试样中单链取代二甲基苄基氯化铵类化合物所属色谱峰;然后将所述试样中单链取代二甲基苄基氯化铵类化合物的色谱峰面积代入步骤(1)所得标准工作曲线计算得所述试样中单链取代二甲基苄基氯化铵类化合物的初始浓度;3)将步骤2)中所得初始浓度代入修正系数矫正得所述试样中单链取代二甲基苄基氯化铵类化合物的最终浓度;所述修正系数k的计算公式为:其中,k是浓度修正系数;为c12-二甲基苄基氯化铵分子的不饱和度;ω待测目标物为所述试样中单链取代二甲基苄基氯化铵类化合物的不饱和度。本发明的方法中,由于单链取代二甲基苄基氯化铵类化合物都含有二甲基苄基氨基官能团,此类化合物的uv特征吸收相似,可比对uv图谱归属定性化合物类别。该类化合物uv在200nm、218nm和263nm处有特征吸收,根据这三个特征吸收峰即可判定待测试样中是否含有单链取代二甲基苄基氯化铵类化合物。一般判断标准为三个所述特征吸收峰的重合度在90%以上。此外,修正系数k是保证测试结果准确性的关键因素。本方法色谱条件的检测器为二极管整列检测器,检测紫外波长为260nm。根据紫外吸收原理,化合物分子的不饱和度是影响化合物紫外吸收的重要因素之一,故化合物分子的不饱和度也是影响本方法色谱峰面积的重要因素之一。本发明归纳总结了单链取代二甲基苄基氯化铵类化合物的紫外吸收强度和分子不饱和度的关系,发现化合物紫外吸收强度与不饱和度成一定比例关系,因此本发明基于上述关系提出用该修正系数矫正其他单链取代二甲基苄基氯化铵类化合物的浓度,从而得出更准确的结果。优选的,步骤1)中所述c12-二甲基苄基氯化铵标准工作曲线浓度范围是10~100mg/l。优选的,步骤2)中所述单链取代的二甲基苄基氯化铵类化合物包括c14-二甲基苄基氯化铵、c16-二甲基苄基氯化铵和/或苄索氯铵。优选的,步骤(2)中对试样进行预处理,步骤为:取样品于25ml容量瓶中,加入20ml乙腈(色谱纯)涡旋震荡1min,超声提取30min,取出,冷却后用乙腈(色谱纯)定容至25ml,混匀,经0.45μm滤膜过滤,得到的滤液即为所述试样。优选的,步骤1)和2)中所述hplc条件为:色谱柱为岛津cn-3柱(250mm×4.6mm×5μm),柱温为25~35℃,进样量为5~20μl,流动相为乙腈+0.1mol/l醋酸铵缓冲溶液(冰醋酸调ph至5.0)=60+40,流速0.5~1.0ml/min,检测波长为260nm。与现有技术相比,本发明具有如下优势:本发明利用uv图谱定性,基于c12-二甲基苄基氯化铵单一对照品对单链取代二甲基苄基氯化铵类化合物进行定量分析,并通过修正系数对定量结果的进行修正,建立了简便、快速、高效、普适性强的检测方法。大大的提高了试验效率,降低了检测成本,拓宽了方法的适用范围。本发明对c14-二甲基苄基氯化铵结果的准确度为83.4%,c16-二甲基苄基氯化铵结果的准确度为83.8%,苄索氯铵的结果准确度为109.3%。本发明是一种简便、快速、高效、普适性强的检测方法,可用于药品原料、化工产品主成分中单链取代二甲基苄基氯化铵类化合物。附图说明图1为本发明实施例中c12-二甲基苄基氯化铵的uv图谱。图2为本发明实施例中c14-二甲基苄基氯化铵样品的uv图谱。图3为本发明实施例中c16-二甲基苄基氯化铵样品的uv图谱。图4为本发明实施例中苄索氯铵样品的uv图谱。具体实施方式下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。实施例中使用的仪器与试剂检测仪器:高效液相色谱仪(岛津lc-20at),超声提取机(jp-120st),电子天平(ja5003n)。实验所用玻璃器皿均用自来水洗净,milli-q水冲洗,烘干后备用。试剂:c12-二甲基苄基氯化铵标准物质(aladdin,99%),c14-二甲基苄基氯化铵(aladdin,98%),c16二甲基苄基氯化铵(aladdin,95%),苄索氯铵(manhage,97.5%),乙腈(色谱纯),milli-q水,醋酸铵(优级纯)。样品为市售c14-二甲基苄基氯化铵、c16二甲基苄基氯化铵和苄索氯铵原料。实施例1(1)样品处理用分析天平称取适量c14-二甲基苄基氯化铵原料,分别配制成浓度约为10mg/l、50mg/l和100mg/l的高中低浓度的系列样品溶液1。用分析天平称取适量c16-二甲基苄基氯化铵原料,分别配制成浓度约为10mg/l、50mg/l和100mg/l的高中低浓度的系列样品溶液2。用分析天平称取适量苄索氯铵原料,分别配制成浓度约为10mg/l、50mg/l和100mg/l的高中低浓度的系列样品溶液3。(2)标准工作溶液的配制用分析天平称取c12-二甲基苄基氯化铵的标准物质,用乙腈配制成100mg/l的储备溶液,然后配制成浓度为10mg/l、30mg/l、50mg/l、80mg/l和100mg/l的标准工作溶液。(3)高效液相色谱法检测色谱条件:色谱柱为岛津cn-3柱(250mm×4.6mm×5μm),柱温为25~35℃,进样量为5~20μl,流动相为乙腈+0.1mol/l醋酸铵缓冲溶液(冰醋酸调ph至5.0)=60+40,流速0.5~1.0ml/min,检测波长为260nm。将(2)中标准系列分别注入高效液相色谱中,根据峰面积和浓度拟合工作曲线,线性范围10~100mg/l,相关系数>0.999。(4)定性鉴定本实施例的标准物质是c12-二甲基苄基氯化铵,先利用hplc建立c12-二甲基苄基氯化铵的标准工作曲线,同时记录c12-二甲基苄基氯化铵的uv图谱。如图1显示,在200nm、218nm和263nm处,c12-二甲基苄基氯化铵有特征吸收峰。该吸收峰为化合物中二甲基苄基氨基官能团不饱和键的特征吸收峰。故认定200nm、218nm和263nm处特征吸收峰为单链取代二甲基苄基氯化铵类化合物的定性uv特征峰。同时,若吸收峰的重合度在90%以上即可定性为单链取代二甲基苄基氯化铵类化合物。图2-4为三个系列样品溶液中特定色谱峰的uv图谱,uv图显示三个待测样品均有200nm、218nm和263nm的特征吸收峰,可以判断出三个系列样品溶液中该特定色谱峰为目标物质峰,且结合产品说明书可得出目标物质为单链取代二甲基苄基氯化铵类化合物。(5)定量分析将待测试样中单链取代二甲基苄基氯化铵的色谱峰面积代入标准工作曲线计算得待测试样中各单链取代二甲基苄基氯化铵的初始浓度。初始浓度再代入修正系数k矫正得最终浓度。在此条件下,c14-二甲基苄基氯化铵、c16-二甲基苄基氯化铵和苄索氯铵的修正因子如表2所示。表2待测单链二甲基苄基氯化铵的浓度修正因子k目标物ukc12-二甲基苄基氯化铵4/c14-二甲基苄基氯化铵41c16-二甲基苄基氯化铵41苄索氯铵82(6)准确度判定:用c14-二甲基苄基氯化铵(aladdin,98%),c16二甲基苄基氯化铵(aladdin,95%),苄索氯铵(manhage,97.5%)配制标准工作溶液绘制曲线。测试供试品溶液中目标化合物的真实浓度,与(5)中理论计算浓度比较,计算本发明的准确度。准确度结果如表3所示:表3系数修正法测试结果准确度结果上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种2,5‑二甲基‑2,4‑己二烯异构体的回收利用方法与流程
- 呢绒面料的后整理工艺的制作方法与工艺
- 使用2‑二甲基氨基‑2‑羟甲基‑1,3‑丙二醇的水溶液从气体混合物中除去酸性气体的方法与流程
- 1,4‑二甲基‑2,5‑二‑1H‑1,2,4‑双三唑硝酸铜二维配合物单晶与应用的制作方法与工艺
- 亲水性纳米复合材料结合质谱分析鉴定糖基化肽段的方法与流程
- 一种粉末活性炭与聚二甲基二烯丙基氯化铵联合混凝的水处理方法与流程
- 聚二烯丙基二甲基氯化铵功能化石墨烯的制备方法与流程
- PVDF球阀的制作方法与工艺
- 5-(2-(8-((2,6-二甲基苄基)氨基)-2,3-二甲基咪唑并[1,2-a]吡啶-6-甲酰胺基)乙氧基)-5-氧代戊酸的固体形式及其制备方法与流程
- 一种季铵化壳聚糖/聚丙烯酸复合纳滤膜及其制备方法与流程