一种同时检测维生素D和维生素K的方法与流程
本发明属于检测技术领域,尤其涉及一种同时检测维生素d和维生素k的方法。
背景技术:
目前,维生素d和维生素k是分开进行检测的,对于维生素d3的检测,样品处理需要4h,且操作复杂繁琐,包括样品皂化、提取、浓缩、洗涤、定容和过滤,样品上机时间为6h,检测一批样品的维生素d3共需10h;对于维生素k2的检测,样品处理需要1.5h,样品上机时间为7h,检测一批样品的维生素k2共需8.5h;完成一批样品的维生素d3和维生素k2检测,共需要18.5h,不利于产品的高效、快速检测。并且,在检测样品维生素d3和维生素k2中,处理过程需要使用大量氢氧化钾、石油醚、乙酸乙酯等对人体伤害较大的试剂,不利于检测人员的健康。
技术实现要素:
有鉴于此,本发明提供了一种同时检测维生素d和维生素k的方法,用于解决现有技术检测维生素d3和维生素k2操作过程繁琐、时间长、不利于检测人员健康的问题。
本发明的具体技术方案如下:
一种同时检测维生素d和维生素k的方法,包括以下步骤:
a)采用正己烷和无水乙醇的混合溶液对待测物进行维生素d3和维生素k2的提取,
b)采用液相色谱法进行所述待测物维生素d3和维生素k2的检测,得到液相色谱数据,根据维生素d3和维生素k2的液相色谱标准工作曲线,得到所述待测物维生素d3和维生素k2的含量。
本发明中,采用正己烷和无水乙醇的混合溶液对待测物进行维生素d3和维生素k2的提取以进行样品处理,避免采用毒性高的溶剂,有利于检测人员的健康,实验结果表明,本发明方法能够同时检测维生素d3和维生素k2,检测的准确率高,采用本发明方法进行维生素d3和维生素k2的检测,每批样品仅需8.5h,大大提高了检测效率。
优选的,所述待测物为钙维生素d维生素k软胶囊。
优选的,所述正己烷和无水乙醇的混合溶液中正己醇和无水乙醇的体积比为(10~20):(80~90)。
更优选的,正己烷和无水乙醇的混合溶液中正己醇和无水乙醇的体积比为20:80。
优选的,所述液相色谱标准工作曲线中维生素d3的浓度为0.128μg/ml~1.28μg/ml;
所述液相色谱标准工作曲线中维生素k2的浓度为0.513μg/ml~5.13μg/ml。
优选的,所述液相色谱法的条件为:
色谱柱为c18色谱柱;
色谱柱柱温为30℃~50℃;
检测波长为264nm;
流动相包括流动相a和流动相b,所述流动相a为95%甲醇-水溶液,所述流动相b为异丙醇;
流动相流速为0.5ml/min~1.5ml/min。
更优选的,色谱柱柱温为40℃;
流动相流速为1ml/min。
优选的,所述c18色谱柱的长度为250nm,所述c18色谱柱的内径为4.6mm,所述c18色谱柱的填料粒径为5μm。
本发明中,c18色谱柱选自agilentc18250×4.6mm,5μm;cnw(上海安谱有限公司)c18250×4.6mm,5μm;迪马(diamonsil)plusc18250×4.6mm,5μm或岛津wondasilc18250×4.6mm,5μm。c18色谱柱优选为agilentc18250×4.6mm,5μm。
优选的,所述液相色谱法采用梯度洗脱。
优选的,所述梯度洗脱中,0.01~20.00min,所述流动相a和所述流动相b的体积比为98:2;20.01~38.00min,所述流动相a和所述流动相b的体积比为50:50;38.01~42.00min,所述流动相a和所述流动相b的体积比为0:100;42.01~45.00min,所述流动相a和所述流动相b的体积比为50:50;45.01~53.00min,所述流动相a和所述流动相b的体积比为98:2。
优选的,所述提取为超声提取。
优选的,所述超声提取的频率为20khz~60khz;
所述超声提取的温度为55℃~65℃;
所述超声提取的时间为25min~35min。
更优选的,超声提取的频率为40khz;
所述超声提取的温度为60℃;
所述超声提取的时间为30min。
本发明中,采用正己烷和无水乙醇的混合溶液对待测物进行维生素d3和维生素k2的提取,提取后进行定容,采用反相色谱法将维生素d3和维生素k2分离,用紫外检测器检测,并用外标法定量,得到待测物中维生素d3和维生素k2的含量。
综上所述,本发明提供了一种同时检测维生素d和维生素k的方法,包括以下步骤:a)采用正己烷和无水乙醇的混合溶液对待测物进行维生素d3和维生素k2的提取,b)采用液相色谱法进行所述待测物维生素d3和维生素k2的检测,得到液相色谱数据,根据维生素d3和维生素k2的液相色谱标准工作曲线,得到所述待测物维生素d3和维生素k2的含量。实验结果表明,本发明方法能够同时检测维生素d3和维生素k2,检测的准确率高,采用本发明方法进行维生素d3和维生素k2的检测,每批样品仅需8.5h,大大提高了检测效率,并且,本发明采用正己烷和无水乙醇的混合溶液对待测物进行维生素d3和维生素k2的提取以进行样品处理,避免了采用毒性高的溶剂,有利于检测人员的健康。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
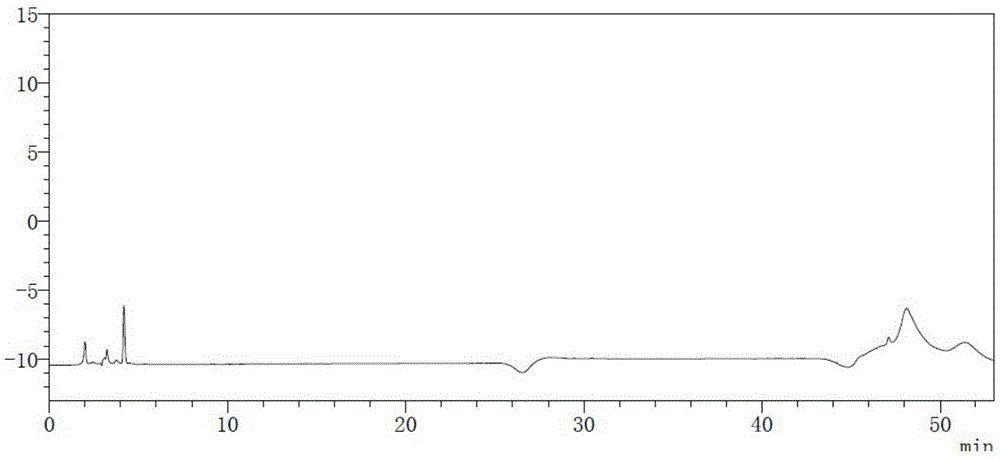
图1为本发明实施例中空白溶液的液相色谱图;
图2为本发明实施例中工作溶液的液相色谱图;
图3为本发明实施例中液相色谱标准工作曲线的维生素d3标准工作曲线;
图4为本发明实施例中液相色谱标准工作曲线的维生素k2标准工作曲线。
具体实施方式
本发明提供了一种同时检测维生素d和维生素k的方法,用于解决现有技术检测维生素d3和维生素k2操作过程繁琐、时间长、不利于检测人员健康的问题。
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
为了进一步理解本发明,下面结合具体实施例对本发明进行详细阐述。
本发明实施例采用的仪器包括高效液相色谱仪(含紫外检测器)、超声仪和电子天平,采用的试剂包括正己烷(ar)、甲醇(色谱纯)、异丙醇(色谱纯)、无水乙醇(ar)、乙酸乙酯(ar)、维生素d3对照品(来源:dr,批号:100656,纯度:99.9%)、维生素k2对照品(来源:usp,批号:r059x0,纯度:99.8%),待测物为汤臣倍健钙维生素d维生素k软胶囊。
实施例1
本实施例进行维生素d3储备液和维生素k2储备液的制备。
维生素d3储备液的制备:精密称取维生素d3标准品3.06mg置于500ml棕色容量瓶中,用无水乙醇溶解,定容,摇匀,即得维生素d3储备液,采用紫外分光光度计测定吸光度,测得浓度为6.4091μg/ml。维生素k2储备液的制备:精密称取维生素k2标准品10.30mg置于100ml棕色容量瓶中,用乙酸乙酯溶解,定容,摇匀,即得维生素k2储备液,采用紫外分光光度计测定吸光度,测得浓度为100μg/ml。维生素d3储备液和维生素k2储备液均放置于-18℃冰箱中储存备用。
实施例2
本实施例进行标准工作溶液的配制。
精密吸取维生素d3储备液10.00ml、维生素k2储备液2.50ml置于25ml棕色容量瓶中,加入无水乙醇定容至刻度,摇匀,即得标准工作溶液,分别精密吸取工作溶液1μl、2μl、5μl、8μl、10μl,在如下液相色谱条件:色谱柱为agilentc18250×4.6mm,5μm;色谱柱柱温为40℃;检测波长为264nm;流动相包括流动相a和流动相b,流动相a为95%甲醇-水溶液,流动相b为异丙醇;流动相流速为1ml/min;采用梯度洗脱,0.01~20.00min,流动相a和流动相b的体积比为98:2;20.01~38.00min,流动相a和流动相b的体积比为50:50;38.01~42.00min,流动相a和流动相b的体积比为0:100;42.01~45.00min流动相a和流动相b的体积比为50:50;45.01~53.00min,流动相a和流动相b的体积比为98:2,进行分析测定,绘制液相色谱标准工作曲线,维生素d3标准工作曲线和维生素k2标准工作曲线请分别参阅图3和图4。其中,液相色谱检测的进样量为20μl。
实施例3
精密称取待测物1.1000g(1g~3g),置于10ml(或25ml)棕色容量瓶中,加入正己烷:无水乙醇(20:80)的混合溶液中,置于60℃超声仪中进行30min超声提取,取出,放置室温,用正己烷:无水乙醇(20:80)的混合溶液定容至刻度,摇匀过0.45μm有机滤膜,得到供试品溶液,进行液相色谱检测的进样量为20μl(10μl~30μl),液相色谱条件条件同实施例2液相色谱条件。
根据公式x=v×c/m×k计算待测物中维生素d3或维生素k2的含量。
公式中:x为待测物中维生素d3或维生素k2含量,单位为μg/粒;
c为供试品溶液中维生素d3或维生素k2的浓度,单位为μg/ml;
m为待测物的质量,单位为g;
v为待测物稀释的体积,单位为ml。
k为单位转换系数。
结果请参阅表1和表2,分别为采用本发明方法检测待测物维生素d3和维生素k2的数据。
表1采用本发明方法检测待测物维生素d3的数据
表2采用本发明方法检测待测物维生素k2的数据
对比例1
根据gb5009.82-2016第四法进行维生素d3的测定,包括以下步骤:
(1)维生素d3标准溶液配制
准确称取维生素d3标准品3.5mg用色谱纯无水乙醇溶解并定容至500ml,得到维生素d3标准储备溶液,于-20℃冰箱中密封保存,有效期3个月。将100μl维生素d3标准储备溶液于10ml的棕色容量瓶中,用无水乙醇定容至刻度,混匀,得到维生素d3标准溶液,分别用1cm石英比色杯,以无水乙醇为空白参比,按公式c维生素d3=a×10000/e计算维生素d3标准溶液的浓度。
公式中:c维生素d3为维生素d3标准溶液的浓度,单位为μg/ml;
a为维生素d3标准溶液的平均紫外吸光值;
10000为换算系数;
e为维生素d31%比色光系数。
(2)待测物的处理
皂化:称取1.0g待测物(准确至0.01g)经均质处理于150ml平底烧瓶中,再加入1.0g抗坏血酸和0.1gbht,混匀,加入30ml无水乙醇,加入10ml~20ml氢氧化钾溶液,边加边振摇,混匀后于恒温磁力搅拌器上80℃回流皂化30min,皂化后立即用冷水冷却至室温,得到皂化液。
提取:将皂化液用30ml水转入250ml的分液漏斗中,加入50ml石油醚,振荡萃取5min,将下层溶液转移至另一250ml的分液漏斗中,加入50ml的石油醚再次萃取,合并醚层。
洗涤:用约150ml水洗涤醚层,重复3次,直至将醚层洗至中性(用ph试纸检测下层溶液ph值),去除下层水相。
浓缩:将洗涤后的醚层经无水硫酸钠(3g)滤入250ml旋转蒸发瓶或氮气浓缩管中,用15ml石油醚冲洗分液漏斗及无水硫酸钠2次,并入蒸发瓶内,并将其接在旋转蒸发器或气体浓缩仪上,于40℃水浴中减压蒸馏或气流浓缩,待瓶中醚剩下2ml时,取下蒸发瓶,氮吹至干,用正己烷定容至10ml,0.22μm有机系滤膜过滤供半制备正相高效液相色谱系统半制备,净化待测液。
反相液相色谱检测的条件如下:
色谱柱:c18柱,柱长250mm,柱内径4.6mm,粒径5μm或具同等性能的色谱柱;
流动相:甲醇+水=95+5;
流速:1ml/min;
检测波长:264nm;
柱温:35℃±1℃;
根据公式x维生素d3=ρ×v维生素d3×f×100/m计算待测物中维生素d3的含量,结果请参阅表3。
公式中:x维生素d3为待测物中维生素d3的含量,单位为μg/粒;
ρ为根据标准工作曲线计算得到的待测物中维生素d3的浓度,单位为μg/ml;
v维生素d3为正己烷定容体积,单位为ml;
f为待测液稀释过程的稀释倍数;
100为待测物中量以每100克计算的换算系数;
m为待测物的称样量,单位为克g。
表3采用gb5009.82-2016第四法检测待测物维生素d3的数据
对比例2
参照tcjc-sop/b-589-2016进行维生素k2的测定,包括以下步骤:
(1)维生素k2标准溶液配制
称取维生素k2标准品5mg精确到0.01mg于50ml容量瓶中,加乙酸乙酯超声溶解,定容,摇匀,过0.45μm的滤膜,即得。
(2)待测物的处理
将20粒待测物取内容物混合均匀并称取约1.5g置于25ml容量瓶中,加入乙酸乙酯,35℃超声(功率500w,频率50khz)处理至提取完全(15min),取出,放冷,定容,摇匀,经0.45μm的滤膜过滤,即得待测液,进行液相色谱检测。
液相色谱检测的条件如下:
色谱柱:以十八烷基硅烷健合硅胶为填充剂(柱长25cm,内径4.6mm,粒径5μm)或同等性能的色谱柱;
柱温:40℃;
检测波长:270nm;
流动相:50%流动相a:甲醇:0.1%乙酸-水溶液=95:5
50%流动相b:异丙醇
流速:1.0ml/min;
根据公式x维生素k2=v维生素k2×c维生素k2/m维生素k2×k维生素k2计算待测物中维生素d3的含量,结果请参阅表4。
式中:x维生素k2为待测物中维生素k2含量,单位为μg/粒;
c维生素k2为供试品溶液中维生素k2的浓度,单位为μg/ml;
m维生素k2为待测物的质量,单位为g;
v维生素k2为待测液稀释的体积,ml。
k维生素k2为单位转换系数。
表4参照tcjc-sop/b-589-2016检测待测物维生素k2的数据
实施例4
该实施例将实施例3检测维生素d3的数据和对比例1检测维生素d3的数据进行对比,将实施例3检测维生素k2的数据和对比例2检测维生素d3的数据进行对比。结果请参阅表5和表6。结果表明本发明检测方法对维生素k2及维生素d3的含量测定科学有效,能对汤臣倍健钙维生素d维生素k软胶囊中维生素k2和维生素d3的含量起到质量控制的目的。
表5实施例3和对比例1检测维生素d3的数据对比
表6实施例3和对比例2检测维生素k2的数据对比
实施例5
本实施例进行方法学验证。
(1)专属性试验
请参阅图2,为本发明实施例中工作溶液按实施例2液相色谱条件测定的液相色谱图,图2表明工作溶液中含有维生素d3峰和维生素k2。不称取样品,按实施例3方法处理空白溶液,按实施例2液相色谱条件测定空白溶液,结果如图1所示,与维生素d3和维生素k2对照品溶液的出峰时间进行对比,结果表明空白溶液在维生素d3和维生素k2的出峰时间处均无吸收峰,表明空白对维生素d3峰和维生素k2的测定结果基本无干扰。
(2)线性范围
实施例2的测试数据分别如表7和表8所示,即维生素d3和维生素k2的测试数据。以峰面积为横坐标,浓度为纵坐标,根据表7和表8的测试数据分别绘制得到维生素d3标准工作曲线和维生素k2标准工作曲线,即图3和图4。结果表明,维生素d3和维生素k2的相关系数r2分别为0.9999206、0.9999592,本发明方法测定维生素d3在浓度0.12818μg/ml至1.28182μg/ml、维生素k2在浓度0.51397μg/ml至5.13970μg/ml之间呈现良好的线性,符合gb/t27404-2008《实验室质量控制规范》的要求(gb/t27404-2008要求相关系数r≥0.99)。
表7维生素d3的测试数据
表8维生素k2的测试数据
(3)检出限和定量限
分析方法的检出限dl和定量限ql由信噪比(s/n)计算。dl定义为s/n=3时对应的待分析浓度,ql定义为s/n=10时对应的待分析浓度。
1)检出限
当信噪比(s/n)为3时,维生素d3和维生素k2的检出限分别为0.01156μg/ml、0.02685μg/ml,当称取量为1.1g时,本发明检测方法中维生素d3和维生素k2检出限分别为10.5072μg/100g、0.2455mg/kg。
2)定量限
当信噪比(s/n)为10时,维生素d3和维生素k2的定量限分别为0.03853μg/ml、0.09000μg/ml,当称取量为1.1g时,本发明检测方法中维生素d3和维生素k2的定量限分别为35.0273μg/100g、0.8182mg/kg。
(4)精密度试验
称取6份待测物,按实施例3方法处理待测物,检测待测物中维生素d3和维生素k2的含量,并计算其rsd(%),维生素d3和维生素k2的数据分别如表9和表10所示。结果表明检测6份待测物维生素d3和维生素k2含量的rsd分别为3.6%、3.3%,本发明检测方法有较好的精密度,符合gb/t27404-2008《实验室质量控制规范》的要求(gb/t27404-2008要求rsd(%)≤7.5%)。
表9精密度试验中维生素d3的数据
表10精密度试验中维生素k2的数据
(5)稳定性试验
将维生素d3、维生素k2供试品溶液分别在室温下放置0h、1h、4h、8h、12h、24h后,按5.1条件测定浓度,计算其rsd(%)。稳定性试验中维生素d3和维生素k2的数据分别如表11和表12所示。结果表明将供试品溶液分别在室温下放置0h、1h、4h、8h、12h、24h后,检测供试品溶液中维生素d3和维生素k2的rsd(%)分别为1.5%、4.3%,表明供试品溶液中维生素d3、维生素k2在室温下24h内的稳定性良好。
表11稳定性试验中维生素d3的数据
表12稳定性试验中维生素k2的数据
(6)准确度试验(回收率)
加标:精密称取约1.1g待测物,待测物中维生素d3和维生素k2含量分别为:5.0μg/g、16μg/g)9份,分成3组,每组3份,各组分别精密加入维生素d3储备液(浓度为6.4091μg/ml)0.60ml、0.80ml、1.00ml;再分别依次精密加入维生素k2储备液(浓度为0.0102794mg/ml)1.10ml、1.60ml、1.90ml,得到加标样。再按实施例3方法处理加标后的待测物。
测得加入量=加标样测得量-待测物测得量
回收率(%)=测得加入量/理论加入量×100%
准确度试验中维生素d3和维生素k2的数据分别如表13和表14所示。
实验结果表明维生素d3和维生素k2的平均回收率分别为97.66%、99.06%,相对标准偏差(rsd)分别为1.7%、1.3%,符合gb/t27404-2008《实验室质量控制规范》的要求(gb/t27404-2008要求回收率为95~105%)。并且,维生素d3检测的准确率得到了提高,提高了2.8%(gb5009.82-2016第四法维生素d3的回收率为95%,本发明检测方法的回收率达97.66%)
表13准确度试验中维生素d3的数据
表14准确度试验中维生素k2的数据
以上结果表明,本发明同时检测维生素d和维生素k的方法的专属性试验、线性范围、精密度试验、稳定性试验、准确度试验、检出限和定量限均符合gb/t27404-2008《实验室质量控制规范》的要求,表明本发明含量测定方法科学有效,能对汤臣倍健钙维生素d维生素k软胶囊中维生素d3和维生素k2含量起到质量控制的目的。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!