一种预处理方法、吗啡的检测方法及应用与流程
本发明涉及复方药物制剂及分析领域,具体涉及一种预处理方法、吗啡的检测方法及应用。
背景技术:
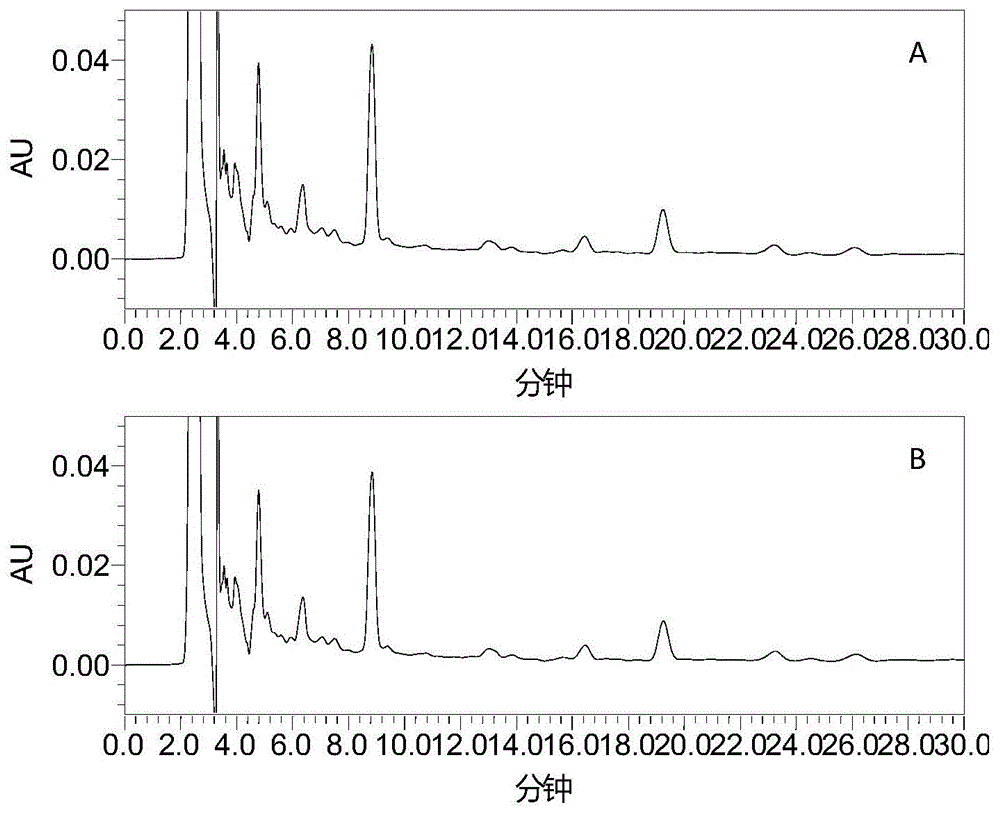
:温肾苏拉甫片收载于《中华人民共和国卫生部药品标准》维吾尔药分册。标准号为ws3-bw-0191-98。本方剂收载于维吾尔族古籍著名药典《卡日巴丁卡德尔》,至今已有1500年的历史。温肾苏拉甫片处方由中亚白及、肉豆蔻、高良姜、附子、肉豆蔻衣、肉桂、罂粟壳、西红花等药材组成。温肾除湿。用于早泄,遗精,遗尿症等。温肾苏拉甫片为多组分复杂体系且含有罂粟壳这种易成瘾的药材,因此评价其质量体系应采取与之相适应的,能提供准确的检测方法,但在温肾苏拉甫片的检测方法中却没有准确的含量测定方法,建立温肾苏拉甫片中含量检测方法能较为全面的反应其所含化学成分的含量,进而对药品质量及安全性进行整体的描述和评价,为控制和保证产品质量的稳定性提供保障。温肾苏拉甫片的治疗效果已经得到临床的验证,并且其效果还在不断发掘,如何能保证药物的质量以及温肾苏拉甫片的有效成分含量,是决定温肾苏拉甫片疗效的基础。目前已经有收载有罂粟壳的薄层鉴别方法以及含量测定方法,但该含量测定方法仅对该罂粟壳药材可以进行测定,而对吗啡含量检测不适用。现有技术中应已经存在对温肾苏拉甫片中罂粟壳的吗啡含量的检测方法,但是该检测方法中待测样品的预处理以及hplc检测过程中仍存在较大问题。例如,石河子大学学报公开了《hplc法测定温肾苏拉甫片中吗啡的含量》,该方法的流动相比例为12.5:75:12.5的甲醇-0.5%醋酸铵(含三乙胺0.08%)-乙腈。重复实施该试验(供试品溶液、对照品溶液以及色谱条件均与之相同)可知,样品中吗啡的色谱峰出现与前后色谱峰均有重叠的情况,重现性差。其待测样品的预处理方法为:“取本品素片50片,研细(过80目筛),精密称取5g,置具塞锥形瓶中,加入碳酸氢钠2g,混合均匀,加氨水溶液18ml,使粉末充分浸润,放置30min,加入乙醚70ml,振揺10min,超声处理(250w,频率20khz)10min,放置过夜,滤过,浸出上清液,残渣再用乙醚超声处理2次,每次60ml,合并提取液,水浴蒸干,残渣用甲醇溶解,定容至10ml量瓶中,并稀释至刻度,摇匀,用微孔滤膜(0.45μm)滤过,即得。”。待测样品的预处理方法存在的问题是:1)加入氨水,浸润较长,由于制剂成粉末状氨水的浸润速度很慢,尤其是在加入乙醚后会出现块状凝结,使样品不成分散体系,无法使样品充分接触溶剂。浸泡过程需要30min左右,整个处理过程需要24h左右。2)多次转移,过程损失较大。3)乙醚具有强挥发性,使用不安全。故该检测方法中待测样品的预处理以及hplc检测过程中仍存在较大问题,由此可导致无法准确测定温肾苏拉甫制剂中的吗啡含量。由于罂粟壳含有吗啡、可待因、罂粟碱等成分等,其中吗啡属于阿片类生物碱,为阿片受体激动剂,具有麻醉镇痛作用,过量可致急性中毒,成人中毒量为60mg,致死量为250mg,具有成瘾性,需要对温肾苏拉甫制剂中的吗啡含量进行测定,确保用药的安全性及药品的疗效。该问题亟待解决。技术实现要素:本发明的目的在于克服现有技术中温肾苏拉甫片中吗啡含量的检测方法中,待测样品的预处理过程中,存在的浸润时间长,样品分散困难无法充分接触溶剂,多次转移、过程损失较大,采用挥发性强溶剂的问题,以及hplc检测过程中存在目标峰与前后色谱峰有重叠的缺陷,而提供一种预处理方法、吗啡的检测方法及应用。本发明通过以下技术方案解决上述技术问题。本发明提供了一种预处理方法,其包括下述步骤:将待测样品和氨的甲醇溶液的混合液,加热回流,滤过后,收集滤液即得供试品溶液;其中,所述待测样品为含有吗啡的药物组合物;所述氨的甲醇溶液中,氨水的体积与所述氨的甲醇溶液总体积之比为(0.10~0.40):1,所述待测样品和所述氨的甲醇溶液的质量体积比为5~15g/50ml。其中,所述预处理方法中,较佳地不添加乙醚。其中,所述加热回流的操作和条件可为本领域常规,一般在具塞锥形瓶中进行。所述加热回流的时间较佳地为0.3~1h,更佳地为0.5h。其中,在所述加热回流的操作前,较佳地将所述混合液称定重量,待加热回流后,冷却,再用所述氨的甲醇溶液补足减失的重量。其中,在所述滤过的操作前,较佳地将加热回流的混合液进行冷却。所述冷却的方式可为本领域常规,一般为自然冷却。所述滤过的操作和条件可为本领域常规,一般采用定量滤纸进行过滤,其目的是除去较大的颗粒物。其中,所述含有吗啡的药物组合物可通过本领域常规方法制得,较佳地通过下述制备方法中的任意一种:方法一:将温肾苏拉甫片去除薄衣后研细,得细粉;方法二:所述含有吗啡的药物组合物的原料包括中亚白及、肉豆蔻、高良姜、附子、肉豆蔻衣、肉桂、罂粟壳和西红花等,可按本领域常规方法制得所述含有吗啡的药物组合物,较佳地通过下述步骤制得:(1)将所述西红花和所述中压白及粉碎,得细粉a;(2)将除所述西红花和所述中压白及以外的原料加水煎煮2~4次(例如3次),每次0.5~1.5h(例如1h),合并煎液,过滤后浓缩成稠膏,加上所述细粉a,混匀即得。方法一中,所述温肾苏拉甫片可为普通的市售药品。所述研细的操作和条件可为本领域常规。所述细粉的粒径可为本领域常规,较佳地为所述细粉全部能通过五号筛,且能通过六号筛的细粉不少于95%。方法二中,一般将方法二制得的含有吗啡的药物组合物按照本领域常规方法制成常规剂型。其中,其中,所述的常规剂型包括以下剂型:片剂、丸剂、散剂、注射剂、酊剂、溶液剂、浸膏剂和软膏剂等,较佳地为片剂。方法二中,所述中亚白及、肉豆蔻、高良姜、附子、肉豆蔻衣、肉桂、罂粟壳和西红花的重量比可为本领域常规,较佳地为20:20:20:10:10:10:10:1。其中,所述含有吗啡的药物组合物中,每克所述药物组合物的活性成分中,含有罂粟壳以吗啡计,不少于204μg。例如,当所述含有吗啡的药物组合物由上述方法一制得时,每克温肾苏拉甫片的活性成分中含吗啡的含量不少于204μg。其中,所述氨的甲醇溶液中,氨水的体积与所述氨的甲醇溶液总体积之比较佳地为0.30:1。其中,所述甲醇可购于fisherchemical,批号为151345。所述氨水可购于四川西陇化工有限公司,批号170323。其中,所述待测样品和所述氨的甲醇溶液的质量体积比较佳地为10g/50ml。若待测样品和氨的甲醇溶液的质量体积比小于5g/50ml,则无法检出吗啡含量或者所得数据不平行(“数据不平行”是指的当处理的两个平行样时,检测结果差距较大,不平行,本发明中所提及的“数据是否平行”均指平行样及其检测结果),若待测样品和氨的甲醇溶液的质量体积比大于15g/50ml,则待测样品中的吗啡无法完全溶解于溶剂中。本发明还提供了一种吗啡的检测方法,其包括下述步骤:将所述供试品溶液经高效液相色谱检测吗啡,即可;其中,色谱条件为:填充剂为十八烷基硅烷键合硅胶;流动相体系包括流动相a、流动相b和流动相c,所述流动相a、所述流动相b和所述流动相c的体积比为(8-10):(81-83):(8-10),所述流动相a为甲醇,所述流动相b为浓度为0.4~0.6%醋酸铵溶液,所述醋酸铵溶液中还包括三乙胺,三乙胺的体积与溶液总体积之比为(0.8~1):1000;,所述流动相c为乙腈。其中,所述供试品溶液进行高效液相色谱检测前,较佳地需采用孔径为0.45μm的有机膜进行过滤。所述有机膜的种类可为本领域常规,例如有机膜可购于天津赛普瑞实验设备有限公司,规格为0.45μm*25mm的有机膜。其中,所述高效液相色谱检测中,其它色谱条件可为本领域常规,较佳地为:进样量为5-25μl;检测波长为287nm;理论塔板数按吗啡峰计算应不低于3000;柱温:40℃;流速:1ml/min。更佳地,所述进样量为20μl。更佳地,所述理论塔板数按吗啡计算应不低于5000。其中,所述流动相a、所述流动相b和所述流动相c的体积比较佳地为9:82:9。其中,所述流动相b较佳地为浓度为0.5%醋酸铵溶液。其中,所述流动相b中,所述醋酸铵溶液中的溶剂可为本领域常规,较佳地为水。其中,所述流动相b中,所述三乙胺的体积与溶液总体积之比较佳地为0.8:1000。本发明还提供了一种所述检测方法在含吗啡药物组合物含量检测中的应用,包含下述步骤:步骤(1)、分别配置一系列浓度的对照品溶液和所述含吗啡的供试品溶液,采用上述检测方法对所述对照品溶液和所述含吗啡的供试品溶液进行分析检测;测得所述对照品溶液和所述含吗啡的供试品溶液中吗啡的峰面积;步骤(2)、根据所述对照品溶液中吗啡的峰面积和相应的浓度,进行线性回归得标准曲线方程;步骤(3)、所述含吗啡的供试品溶液中吗啡的含量由步骤(1)中测得的峰面积的代入步骤(2)中得到的标准曲线方程,即可。所述步骤(1)中,所述对照品溶液或所述供试品溶液的配制可采用能将其溶解的常规溶剂,例如氨的甲醇溶液,所述氨的甲醇溶液中,氨水的体积与所述氨的甲醇溶液总体积之比为(0.10~0.40):1,较佳地为0.30:1。所述对照品溶液的浓度可参考本领域制作浓度与峰面积的标准曲线方程时的浓度,例如20~130μg/ml;所述供试品溶液的浓度可根据对照品浓度的范围进行选择,例如30mg/ml。所述步骤(2)中,所述标准曲线方程的获得可参考本领域的常规计算方法,例如,以所述对照品溶液中吗啡的浓度为横坐标,以相应吗啡浓度对应的峰面积为纵坐标,进行线性回归得标准曲线方程。所述检测方法在含吗啡药物组合物含量检测中的应用,可包含以下步骤:(1)称取吗啡对照品(例如购于中国药品生物制品检定所,批号171201-201123)0.02116g,置于100ml容量瓶中,以30%氨的甲醇溶液作溶剂溶解并稀释至刻度,得到吗啡的总浓度为205.464μg/ml的溶液,作为吗啡对照品储备溶液;从吗啡对照品储备溶液中取适量分别置于容量瓶中,配制成吗啡浓度分别为20.546μg/ml、41.929μg/ml、61.639μg/ml、82.185μg/ml、102.732μg/ml、123.278μg/ml的对照品溶液;取含吗啡的药物组合物10g左右,精密称定,置具塞锥形瓶中,精密加入氨甲醇溶液(氨水的体积与氨的甲醇溶液总体积之比为0.3:1)50ml,密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用氨甲醇溶液(氨水的体积与氨的甲醇溶液总体积之比为0.3:1)补足减失的重量,滤过,得供试品溶液;(2)取所述对照品溶液和供试品溶液各20μl进样,采用上述分析检测方法,根据所述对照品溶液中吗啡的峰面积和相应的溶液质量浓度,进行线性回归得标准曲线方程;(3)所述供试品溶液中吗啡的含量由测得的峰面积的代入标准曲线方程计算得相应的吗啡浓度。在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。本发明所用试剂和原料均市售可得。本发明的积极进步效果在于:(1)本发明待测样品的预处理方法有利于待测样品均匀分散;快速(处理过程时间短不足1h,无需过夜),处理过程简单,提取率高;经济;安全(例如可采用常用溶剂甲醇,可无需使用乙醚);能够有效地提取出待测组分,有益于待测样品的准确、快速的检测,确定了吗啡的含量限度,从而能保证药品的安全性、有效性及质量均一性。(2)本发明的检测方法可用于检测温肾苏拉甫制剂中吗啡含量,吗啡的峰形和分离度良好,色谱峰单一,不存在包峰现象,且检测方法快速,简单,精密度高,重复性好,准确性高,可准确控制产品质量。附图说明图1为对比例1的供试品溶液的hplc图,其中,图1a、图1b表示供试品溶液平行进样两次。图2为实施例2的供试品溶液的hplc图,其中,图2a、图2b表示供试品溶液平行进样两次。图3对比例2、实施例3~5供试品溶液的hplc图,其中,图3a~图3d分别为对比例2、实施例3、实施例4、实施例5的供试品溶液的hplc图。图4为实施例6、7的供试品溶液的hplc图,其中,图4a为实施例6供试品溶液的hplc图,图4b为实施例7供试品溶液的hplc图。图5为实施例7、对比例3的供试品溶液和对照品溶液中吗啡检测的hplc图,其中,图5a为对比例3的供试品溶液和对照品溶液中吗啡检测的hplc图,图5b为实施例7的供试品溶液和对照品溶液中吗啡检测的hplc图。图6为空白组、吗啡对照品、实施例7供试品溶液和缺罂粟壳阴性样品的hplc图,其中,图6a为空白组(30%氨的甲醇溶液),图6b为吗啡对照品(吗啡浓度为30μg/ml的混合溶液,溶剂为30%氨的甲醇溶液),图6c为实施例7供试品溶液,图6d为缺罂粟壳阴性样品。图7为实施例7的供试品溶液在不同柱温下检测吗啡的hplc图,其中,图7a的柱温为25℃,图7b的柱温为30℃,图7c的柱温为35℃,图7d的柱温为40℃。图8为实施例7的供试品溶液在不同品牌柱子下检测吗啡的hplc图,其中,图8a色谱柱为agilenteclipseplusc18(4.6×250mm,5μm),图8b色谱柱为sunfiretmc18(4.6×250mm,5μm),图8c色谱柱为wondasilr(4.6×250mm,5μm)。具体实施方式下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。若无特殊说明,下述实施例和对比例中采用的实验材料均如下所示:含有吗啡的药物组合物均通过下述步骤制得:将温肾苏拉甫片(生产厂家:新疆维吾尔药业有限责任公司,批号为917060)去除薄衣后,研细,得细粉,所得细粉全部能通过五号筛,且能通过六号筛的细粉不少于95%。研细的操作和条件可为本领域常规;色谱甲醇购于fisherchemical,批号151345;氨水购于四川西陇化工有限公司,批号170323;吗啡对照品购于中国药品生物制品检定所,批号171201-201123;有机模购于天津赛普瑞实验设备有限公司,规格为0.45μm*25mm。在图1~8中,纵坐标a.u代表吸光度。实施例1标准曲线的建立与线性范围吗啡对照品储备溶液制备:称取吗啡对照品0.02116g,置于100ml容量瓶中,以30%氨的甲醇溶液作溶剂溶解并稀释至刻度,得到吗啡的总浓度为205.464μg/ml的溶液。精密吸取吗啡对照品储备溶液适量,依次用30%氨的甲醇溶液(30%氨的甲醇溶液配制过程:量取氨水300ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得)逐级稀释成标准工作液,标准工作液中,吗啡浓度分别为20.546μg/ml、41.929μg/ml、61.639μg/ml、82.185μg/ml、102.732μg/ml、123.278μg/ml。进行高效液相色谱检测,制得标准曲线。高效液相色谱法检测的条件为:流动相体系包括流动相a、流动相b和流动相c,流动相a、流动相b和流动相c的体积比为9:82:9,流动相a为甲醇,流动相b为浓度为0.5%醋酸铵溶液(含三乙胺的体积与溶液总体积之比0.08%),流动相c为乙腈。1000ml流动相b的配制过程:精密称取醋酸铵5g至1000ml容量瓶中,用1ml移液管量取三乙胺0.8ml至相同容量瓶中加入纯化水溶解定容至刻度即得。进样量:20μl检测波长:287nm理论塔板数按吗啡峰计算应不低于5000柱温:40℃流速:1ml/min色谱柱agilent(250mm×4.6mm,5μm)色谱柱中填充剂:十八烷基硅烷键合硅胶以标准工作液进样浓度(x)为横坐标,峰面积积分值(y)为纵坐标,绘制吗啡的标准溶液工作曲线,进行线性回归得标准曲线方程及其相关系数:y=5900x-21700,相关系数r=0.9993;结果表明吗啡对照品在20.516~123.278μg/ml范围内线性关系良好。表1为吗啡标准曲线数据。表1吗啡标准曲线数据浓度μg·ml-120.54641.92961.63982.185102.732123.278峰面积105207220882343151460223576901713670对比例1(供试品溶液的提取方式不同)供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入20%氨的甲醇溶液50ml(20%氨的甲醇溶液配制过程:量取氨水200ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得),密塞,称定重量,超声0.5小时,放冷,再称定重量,用20%氨的甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得;对照品溶液的制备:取吗啡对照品适量,精密称定,加甲醇溶液,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,进行高效液相色谱检测,具体的检测方法和条件均与实施例1相同。图1为对比例1的供试品溶液的hplc图,其中,图1a、图1b表示供试品溶液平行进样两次。实施例2供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入20%氨的甲醇溶液50ml(20%氨的甲醇溶液配制过程:量取氨水200ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得),密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用20%氨的甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得;对照品溶液的制备:取吗啡对照品适量,精密称定,加甲醇溶液,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,进行高效液相色谱检测,具体的检测方法和条件均与实施例1相同。图2为实施例2的供试品溶液的hplc图,其中,图2a、图2b表示供试品溶液平行进样两次。表2为对比例1与实施例2的供试品溶液在保留时间、面积和usp理论塔板数的数据对比。表2提取方式保留时间min面积au*minusp理论塔板数对比例1超声平行样119.2219570.613164.8对比例1超声平行样219.3199709.512552.9实施例2回流平行样119.3266779.413329.4实施例2回流平行样219.3270362.612788.9由表2、图1、图2可知,对比例1超声提取峰面积比回流提取小,含量低,提取不完全,因此选择实施例2回流的方式作为供试品提取方法。对比例2(供试品溶液中溶剂种类为甲醇)供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;对照品溶液的制备:取吗啡对照品适量,精密称定,加甲醇,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,色谱条件与系统实用性试验,注入高效液相色谱仪,进行高效液相色谱检测,具体的检测方法和条件均与实施例1相同。记录时间应不少于30分钟。实施例3供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入20%氨的甲醇溶液50ml(20%氨的甲醇溶液配制过程:量取氨水200ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得),密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用20%氨的甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得;对照品溶液的制备:取吗啡对照品适量,精密称定,加甲醇,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,色谱条件与系统实用性试验,注入高效液相色谱仪,进行高效液相色谱检测,具体的检测方法和条件均与实施例1相同。记录时间应不少于30分钟。实施例4供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入30%氨的甲醇溶液50ml(30%氨的甲醇溶液配制过程:量取氨水300ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得),密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用30%氨的甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得。对照品溶液的制备:取吗啡对照品适量,精密称定,加甲醇,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,进行高效液相色谱检测,具体的检测方法和条件均与实施例1相同。记录时间应不少于30分钟。实施例5供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入40%氨的甲醇溶液50ml(40%氨的甲醇溶液配制过程:量取氨水400ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得),密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用40%氨的甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得;对照品溶液的制备:取吗啡对照品适量,精密称定,加甲醇,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,进行高效液相色谱检测,具体的检测方法和条件均与实施例1相同。记录时间应不少于30分钟。图3对比例2、实施例3~5供试品溶液的hplc图,其中,图3a~图3d分别为对比例2、实施例3、实施例4、实施例5的供试品溶液的hplc图。表3为对比例2、实施例3~5中不同溶剂种类或不同溶剂浓度对峰面积的影响表3由表3、图3可知,吗啡含量受氨浓度影响,在30%及40%氨甲醇溶液中稳定,40%氨甲醇溶液中吗啡峰面积(含量)增加不多,因此选择30%氨甲醇溶液作为提取溶剂。实施例6供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入30%氨的甲醇溶液50ml(30%氨的甲醇溶液配制过程:量取氨水300ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得),密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用30%氨的甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得;对照品溶液的制备:取吗啡对照品适量,精密称定,加体积比为30%氨的甲醇溶液,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,色谱条件与系统实用性试验:以十八烷基硅烷键合硅胶为填充剂,色谱柱agilent(250mm×4.6mm,5μm);以体积比9.5:81:9.5的甲醇-浓度为0.5%醋酸铵溶液(含三乙胺的体积与溶液总体积之比为0.08%)-乙腈为流动相;检测波长为287nm;理论塔板数按吗啡计算应不低于5000;记录时间应不少于30分钟。实施例7供试品溶液的制备:取含有吗啡的药物组合物10g,精密称定,置具塞锥形瓶中,精密加入30%氨的甲醇溶液50ml(30%氨的甲醇溶液配制过程:量取氨水300ml至1000ml容量瓶中,加入甲醇,混匀,定容至刻度即得),密塞,称定重量,加热回流0.5小时,放冷,再称定重量,用30%氨的甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得;对照品溶液的制备:取吗啡对照品适量,精密称定,加体积比为30%氨的甲醇溶液,制成每1ml含吗啡30μg的混合溶液,作为对照品溶液;测定:分别精密吸取对照品溶液与供试品溶液各20μl,注入高效液相色谱仪,色谱条件与系统实用性试验:以十八烷基硅烷键合硅胶为填充剂,色谱柱agilent(250mm×4.6mm,5μm);以体积比9:82:9的甲醇-0.5%醋酸铵(含三乙胺0.08%)-乙腈为流动相;检测波长为287nm;理论塔板数按吗啡计算应不低于5000;记录时间应不少于30分钟。图4为实施例6、7的供试品溶液的hplc图,其中,图4a为实施例6供试品溶液的hplc图,图4b为实施例7供试品溶液的hplc图。表4为实施例6、7中不同流动相比例对峰面积的影响。表4由表4、图4可知,实施例7中流动相比例为9:82:9峰型及分离效果较好。对比例3石河子大学学报(自然科学版)2008年4月第26卷第2期公开了《hplc法测定温肾苏拉甫片中吗啡的含量》,重复实施其实验方法(供试品溶液、对照品溶液以及色谱条件均与之相同,详见公开的第2部分实验方法)。图5a为对比例3公开的供试品溶液和对照品溶液中吗啡检测的hplc图。由图5a可知,样品中吗啡的色谱峰出现(出峰时间为9.4min)与前后色谱峰均有重叠的情况,重现性差。图5b为实施例7公开的供试品溶液和对照品溶液中吗啡检测的hplc图。由图5b可知,样品中吗啡的色谱峰出现(出峰时间为27.2min)单一,不存在包峰现象,重现性好。效果实施例1专属性试验:在上述条件下对空白溶液、供试品溶液、对照品溶液及阴性样品溶液分别进行hplc分析,结果显示表明阴性样品溶液在吗啡对照品色谱峰处无干扰峰出现,处方中其他成分对测定无影响,结果见附图4。图6为空白组、吗啡对照品、实施例7供试品溶液和缺罂粟壳阴性样品的hplc图,其中,图6a为空白组(30%氨的甲醇溶液),图6b为吗啡对照品(吗啡浓度为30μg/ml的混合溶液,溶剂为30%氨的甲醇溶液),图6c为实施例7供试品溶液,图6d为缺罂粟壳阴性样品。图6d中,缺罂粟壳阴性样品:原料以中亚白及、肉豆蔻、高良姜、附子、肉豆蔻衣、肉桂和西红花的重量比为20:20:20:10:10:10:1,通过下述步骤制得阴性样品:(1)将西红花和中压白及粉碎,得细粉;(2)将除西红花和中压白及以外的原料加水煎煮3次,每次1h,合并煎液,过滤后浓缩成稠膏,加上步骤(1)所得细粉,混匀后,即得物料a,将物料a和30%氨的甲醇溶液(物料a和氨的甲醇溶液的质量体积比为10g/50ml)的混合液,加热回流,滤过后,收集滤液即为“缺罂粟壳阴性样品。测定法:分别精密吸取空白组、吗啡对照品溶液、供试品溶液及阴性样品溶液各20μl,注入高效液相色谱仪,进行高效液相色谱检测,具体的检测方法和条件均与实施例1相同。记录时间应不少于30分钟。图6a~6d的实验条件,除进样溶液不一致外,其它测试条件均相同。效果实施例2检测限的测定:吸取浓度为1.23μg/ml的吗啡溶液,进hplc分析,具体的测试条件与实施例1相同,进样量20μl。按信噪比(s/n)为3时计算,吗啡的检测限为1.23μg/ml。定量限的检测:吸取浓度为2.47μg/ml的吗啡溶液,进hplc分析,具体的测试条件与实施例1相同,进样量20μl。按信噪比(s/n)为10时计算,吗啡对照品的定量限为2.47μg/ml。效果实施例3精密度的测定供试品溶液的制备:除称取重量为10.0061g外,其它操作和条件与实施例2中供试品溶液的制备方法均相同。进样量20μl,重复进样6次,具体的测试条件与实施例1相同,测定峰面积,计算得知rsd小于8%,表明其仪器精密度良好。数据见表5。连续两天测试,计算日间精密度结果,得知rsd小于8%,表明其日间精密度良好,数据见表6。rsd为相对标准偏差(relativestandarddeviation),指标准偏差与计算结果算术平均值的比值。表5吗啡仪器精密度测定结果(n=6)表6吗啡日间精密度测定结果效果实施例4重复性试验供试品溶液的制备:除称取重量为10g、温肾苏拉甫片(生产厂家:新疆维吾尔药业有限责任公司批号9170601)外,其它操作和条件与实施例2中供试品溶液的制备方法均相同。分别制备6上述供试品溶液,进样20μl,具体的测试条件与实施例1相同,记录色谱峰的峰面积,计算含量,样品中吗啡的rsd为0.84%,说明方法重复性良好。结果见表7。表7重复性试验结果效果实施例5稳定性试验供试品溶液的制备:除称取重量为10g、温肾苏拉甫片(生产厂家:新疆维吾尔药业有限责任公司批号9170601)外,其它操作和条件与实施例2中供试品溶液的制备方法均相同。供试品溶液配制完成后,分别在配制完成后的0、1、3、5、7、9、11、24、36和48h各进样20μl,结果吗啡含量的rsd为1.98%,表明供试品溶液在48h内稳定。结果见表8。表8稳定性测定结果效果实施例6耐用性测试采用agilent(250mm×4.6mm,5μm)色谱柱,检测波长为287nm,进样量为20μl,柱温为40℃时,在流速为1ml/min时,分别在温度为25℃、30℃、35℃和40℃,采集30分钟内的色谱,具体的检测方法和条件均与实施例1相同。表9为实施例7的供试品溶液在不同柱温下吗啡的保留时间和usp理论塔板数。图7为实施例7的供试品溶液在不同柱温下检测吗啡的hplc图,其中,图7a的柱温为25℃,图7b的柱温为30℃,图7c的柱温为35℃,图7d的柱温为40℃。图7a~图7d的实验条件,除柱温不一致外,其它测试条件均相同。表9不同柱温出峰时间色谱柱温度℃保留时间usp理论塔板数2526.41410476.153020.50713316.263520.49413697.034018.28112757.97由表9、图7可知,在温度为25℃、30℃、35℃和40℃,测定的结果对吗啡的分离度和对称性等均无影响,该温度范围内均可采用进行吗啡含量测定。在流速为1ml/min时,温度为40℃,分别用不同品牌的柱子考察温肾苏拉甫片吗啡的分离度和对称性,采集30分钟内的色谱,具体的检测方法和条件均与实施例1相同。表10为实施例7的供试品溶液在不同色谱柱下吗啡的保留时间和usp理论塔板数。图8为实施例7的供试品溶液在不同品牌柱子下检测吗啡的hplc图,其中,图8a色谱柱为agilenteclipseplusc18(4.6×250mm,5μm),图8b色谱柱为sunfiretmc18(4.6×250mm,5μm),图8c色谱柱为wondasilr(4.6×250mm,5μm)。图8a~图8d的实验条件,除色谱柱种类不一致外,其它测试条件均相同。表10色谱柱保留时间usp理论塔板数agilenteclipseplusc18(4.6×250mm,5μm)18.28112733.13sunfiretmc1823.95310777.51wondasilr26.36911819.52由表10、图8可知,用不同品牌的柱子考察温肾苏拉甫片吗啡的分离度和对称性,结果对吗啡的分离度和对称性等均无影响,该方法适用性高。效果实施例7加样回收率试验取吗啡对照品适量,精密称定,加30%氨的甲醇溶液溶解,制成吗啡浓度为544.958μg/ml。取已准确进行含量测定(批号:9170601,吗啡含量为246.8μg/g)的约5g,精密称定,置具塞锥形瓶中,分别加入浓度为544.958ug/ml的吗啡对照品1ml、1ml、1ml、2ml、2ml、2ml、3ml、3ml、3ml,再用50ml移液管精精密加入49ml、49ml、49ml、48ml、48ml、48ml、47ml、47ml、47ml,总计溶液体积为50ml。按供试品溶液制备方法制备溶液,进样20μl,测定,记录峰面积。计算加样回收率,回收率在94-103%之间,结果表明回收率良好。吗啡平均回收率为99.20%,rsd为3.65%。结果见表11。回收率%=(c-a)/b×100%式中a为供试品所含被测成分量;b为加入对照品量;c为实测值。表11吗啡加样回收率(n=9)效果实施例8供试品溶液的制备:除称取重量为1g、温肾苏拉甫片(生产厂家:新疆维吾尔药业有限责任公司)(下述15个批号)外,其它的具体检测方法和条件均与实施例7相同。每批平行2份。分别精密吸取实施例7的对照品溶液和上述供试品溶液各20μl,注入液相色谱仪,依次测定各批样品。计算吗啡的含量,结果见表12。表1215批样品测定(n=15)编号批号含量μg/g11610023862161159439314120632341703084655171154147761703075467170306528817115516399170710119710170709122411170118255121702371113131610014101416115846815140644402本发明温肾苏拉甫片中吗啡的含量限度的制定:15批次制剂中吗啡含量范围为255-1639μg/g,平均为725μg/g;由于不同批次吗啡含量差异较大(可能为不同批次药材受产地及采收时间的影响造成含量变化),取最低值下浮20%作为含量限度,暂定每克温肾苏拉甫片中含吗啡不得低于204μg/g(106μg/片)(0.02%)(其中,“106μg/片”是指温肾苏拉甫片的每片含有吗啡106μg;“0.02%”是指该片剂的百分含量,百分含量%=0.000204g/1g*100=0.02%)当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1