一种手性分子纯度弱测量检测方法及其装置与流程
本发明属于利用光学手段进行检测的技术领域,具体地,涉及一种手性分子纯度弱测量检测方法及其装置。
背景技术:
手性是广泛存在于自然界的本质属性之一,手性对映体分子量和官能团相同,空间结构镜像对称。手性药物的药效作用与它的构型密切相关,不同构型的对映体进入人体后往往产生不同甚至相反的药理学活性,例如:抗结核药乙胺丁醇,其另一个对映体可能导致失明;利尿剂依托唑啉,有效构型利尿,另一构型竟是抑尿;苯并吗啡烷作为一种镇定药,有效构型无成瘾性,而另一构型有高度成瘾性且效果减弱。而在已知的医药工业中,约有30%~40%的药物是手性的,只有约20%的手性药物是以纯的对映体形式销售。因此,手性分子纯度的检测具有重要意义。
手性分子纯度的检测方法有多种,如色谱法,圆二色谱法,核磁共振法,传感器法等。常见的色谱法有气相色谱、液相色谱,通过手性选择剂与两种对映体结合能力的差异将手性对映体分离。该方法能进行精确定量定性地检测手性分子,但是价格昂贵,操作复杂,需要特定的手性选择剂标记;圆二色谱法利用的是手性分子对左右旋圆偏振光吸收和传播速度的差异。其优点是选择性好,不受杂质的干扰,无损,但是要求在紫外波段有吸收,信号较弱,采集时间长;磁共振法利用的是手性选择剂使异构体的化学环境不等价,从而在nmr谱图上产生不同的化学位移及峰面积。该方法能够直接获得对映体混合物纯度,然而仪器昂贵,操作复杂需要手性选择剂。
上述方法各有优缺点,因此在本技术领域发展一种高灵敏度,价格低廉,操作简单的手性分子纯度检测方法具有重要意义。
技术实现要素:
本发明的目的在于提供一种手性分子纯度弱测量检测方法及其装置,以便于为手性分子纯度检测提供高灵敏度、价格低廉、操作简单的方法和装置。
为此,本发明公开一种手性分子纯度弱测量检测方法,包括下列步骤;
s1.入射光通过所述第一线性偏振片后,直线前进经过装有待测样品溶液的样品池,再透过所述四分之一波片与所述第二线性偏振片后出射到光谱仪,其中所述第一线性偏振片和所述样品池用于选择系统的前选择态,所述第二线性偏振片与所述四分之一波片用于选择系统的后选择态;在频域弱测量中,经过所述第二线偏振片的投影测量后得到放大的弱值,使得出射光谱的中心波长产生移动;
s2.通过所述光谱仪记录所述出射光通过样品池前后的光谱中心波长移动;
s3.根据所述光谱中心波长移动计算所述待测样品溶液的旋光度的大小;
s4.测量待测样品溶液的浓度;
s5.配制与待测样品溶液相同浓度的左旋纯对映体溶液;将上述的相同浓度的左旋纯对映体溶液替换待测样品溶液并重复上述步骤s1.~s3.,得到所述左旋纯对映体溶液的旋光度的大小;
s6.所述步骤s3.得到的所述待测样品旋光度与所述步骤s5.得到的所述纯对映体的旋光度之比即为手性对映体纯度。
进一步地,所述前选择态为:
进一步地,所述出射光谱的中心波长产生移动为:
进一步地,所述β为0.02。在该系统中当β值为0.02,α小于0.025rad时,中心波长的偏移与样品的旋光度成线性关系。
进一步地,步骤s4.测量待测样品的浓度优选紫外分光光度计测量所述待测样品浓度的方法,还可以用其他测量浓度的方法代替。
另一方面,本发明还公开了一种手性分子纯度弱测量检测装置,包括旋光度检测装置和浓度检测装置,所述旋光度检测装置包括光源、第一线性偏振片、样品池、四分之一波片、第二线性偏振片和用于记录光谱中心波长移动的光谱仪;所述第一线性偏振片、样品池、四分之一波片和第二线性偏振片沿光路依次设置。所述浓度检测装置可以为紫外分光光度计。
进一步地,所述光源为超辐射发光二极管,光源频谱为高斯分布。
进一步地,所述第一线性偏振片的透射轴与竖直方向的夹角为π/4;所述第二线性偏振片的透射轴与第一线性偏振片的透射轴垂直。所述第一线性偏振片用来进行前选择。
进一步地,所述样品池为透明容器。
进一步地,所述四分之一波片相对第二线性偏振片旋转的角度为β,β为0.02。四分之一波片与第二线性偏振片共同构成后选择态。
本发明发展了一种基于量子弱测量原理的手性对映体混合物的纯度检测方法,该方法灵敏度高,无需分离样品,仪器结构简单,可实时监测,在药品生产中有巨大的应用潜力。
附图说明
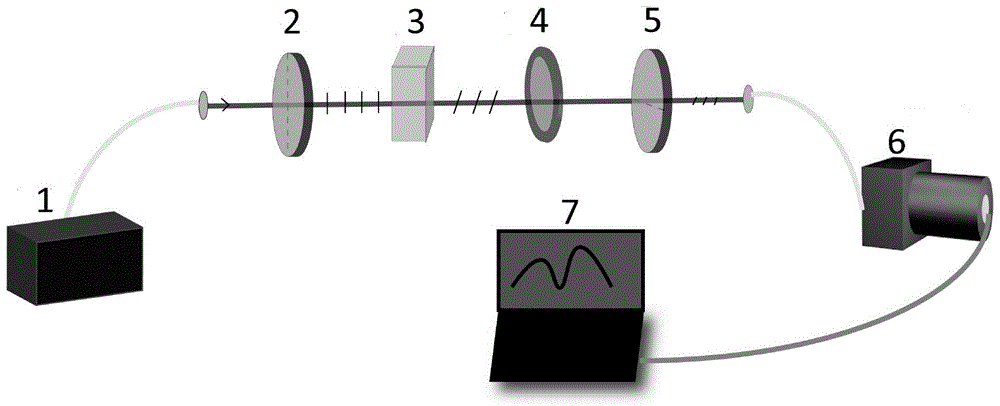
图1手性分子纯度弱测量检测装置结构示意图。其中,1、光源,2、第一线性偏振片,3、样品池,4、四分之一波片,5、第二线性偏振片,6、光谱仪,7、电脑。
图2固定β值为0.02时,光谱中心波长的偏移随α变化的响应图像。
图3(a)混合脯氨酸溶液紫外吸收光谱强度分布图。(b)混合脯氨酸溶液浓度与紫外强度的响应图像。
图4(a)总浓度4g/l的不同比例脯氨酸对映体混合物的中心波长移动实时采集图像,图中(1)至(5)对应的样品为不同比例的脯氨酸对映体混合物,其左右旋的比例分别为0:4,1:3,2:2,3:1,4:0。(b)出射光谱形状对不同纯度脯氨酸对映体浓度的响应,(1)至(5)分别对应(a)中相应表示对映体混合物的比例。
图5不同浓度对映体混合物中心波长移动实时采集数据经平滑消噪后的图像,(a)至(d)混合脯氨酸对映体溶液的总浓度分别为4g/l,2g/l,1g/l,0.5g/l。
图6不同左右旋比例的脯氨酸对映体混合溶液中心波长偏移随浓度变化的响应曲线。
图7偏光仪出射光强随脯氨酸溶液浓度变化的响应图像。
具体实施方式
以下通过下面的实施例更详细地描述本发明,但是,这些实施例并不限制本发明的范围。
材料与方法
1实验试剂
d-脯氨酸(c5h9no2)、l-脯氨酸(c5h9no2)购于美伦生物公司(纯度均>99%)、市售去离子水;
紫外-可见分光光度计品牌:日本岛津uv-2700;附件:电脑,六联自动样器池架及石英比色皿。
实施例1:手性分子纯度弱测量检测装置
一种手性分子弱测量检测装置,采用直线型共路弱测量系统,包括光源、第一线性偏振片、样品池、四分之一波片、第二线性偏振片和光谱仪。
在这个系统中,光源为超辐射发光二极管(sld,thorlabsinc.,sld830s-a20),中心波长为830nm,带宽40nm,用于提供一个高斯光谱的指针态。第一线性偏振片p1(thorlabsinc.,lpvis050-mp,消光比:100000:1)用来进行前选择,其透射轴与竖直方向的夹角为π/4,在系统中安装长度为2cm的比色皿作为样品池(sc)。四分之一波片(qwp,thorlabsinc.,wpmq05m-830)qwp相对第二线性偏振片旋转的角度为β,与第二线性偏振片共同构成后选择态。第二线性偏振片透射轴与竖直方向的夹角为-π/4。输出光束由光谱仪(海洋光学hr4000)接收,光谱仪与计算机连接,用软件oceanview进行光谱分析,设置软件的数据保存间隔时间为100毫秒。
经过第一线性偏振片选择后的前选择态表示为:
其中α表示样品的旋光度。
经过第二线性偏振片后,后选择态为:
其中β表示qwp相对第二线性偏振片旋转的角度。系统中的可观测算符为
a=|r><r|-|l><l|(3)
结合弱值定义式
其中
出射光谱中心波长移动为:
式子中λ0是光源的中心波长830nm,δλ为高斯指针的带宽40nm。由式(6)可知中心波长的偏移与样品的旋光度a以及qwp的指向角β密切相关,在该系统中当β值较小时,在一定的角度范围内,中心波长的偏移与样品的旋光度成线性关系。因此,通过这个线性区间,我们能实现对旋光度的测定。
通过实验得到的旋光度与中心波长的偏移,我们模拟出β值为0.02。因此图2为β值设置成0.02时,出射光谱中心波长的偏移随α变化的响应图像。从图2可以看出,当α角小于0.025rad时,α角与中央波长的位移具有良好的线性关系。
手性样品产生的旋光度可以由公式(7)计算
[α]=100α/l·c(7)
式(7)中c为手性样品溶液的浓度(单位为g/100ml),l为光通过样品的长度(单位为dm),α为样品溶液的旋光度,[α]为比旋光度。
对于总浓度为c,具有不同比例的手性对映体混合物,假设左旋对映体所占比例为a(0≤a≤1),则右旋所占比例为1-a。部分消旋后,对旋光度有贡献的净手性分子占比为2a-1,其浓度为(2a-1)×c,根据公式(7)可知其产生的旋光度为
则
[α]mix就是消旋后对映体混合物的比旋光度,[α]pure是纯对映体的比旋光度,
本实施例中,光源为,sld:超辐射发光二极管;p1是指,第一线性偏振片;sc是指,样品池;qwp是指,四分之一波片;p2是指,第二线性偏振片。
实施例2:手性分子纯度弱测量检测方法
配制了五组不同左右旋比例的脯氨酸溶液,其比例分别为4:0、3:1、2:2、1:3、0:4,每组溶液含有四个总浓度梯度:0.5g/l、1g/l、2g/l、4g/l。
2.1紫外分光光度法测量脯氨酸对映体混合物的浓度
测量所有样品的紫外吸收光谱,每个样品重复测量三次。数据处理时,选取波长205-230nm范围内光谱曲线下的积分面积作为紫外吸收强度。图3(a)是五组不同比例脯氨酸对映体紫外吸收光谱强度分布图,脯氨酸溶液左右旋对映体的比例(1)至(5)组分别为4:0、3:1、2:2、1:3、0:4,从图中可以看出,总浓度相同的不同比例左右旋脯氨酸混合溶液的紫外吸收强度相同,说明d-脯氨酸,l-脯氨酸具有相同的紫外光吸收能力。图3(b)展现了d-脯氨酸对映体紫外吸收强度随浓度变化响应图像,从图中可以看出,紫外光的吸收强度与脯氨酸浓度有良好的线性关系。
2.2脯氨酸溶液的旋光度检测
在实施例1所述直线共路弱测量系统用于检测手性分子的装置中,首先将7毫升的去离子水加入到样品池中,通过调节前后偏振片来选择合适的初始工作点。待系统稳定后,将样品池中去离子水换成7毫升的待测溶液,稳定测量后,更换下一组待测溶液,重复这个操作。图4(a)显示了总浓度4g/l不同比例脯氨酸对映体混合物的中心波长偏移实时采集图像,(1)到(5)表示5种不同左右旋比例,分别为0:4,1:3,2:2,3:1,4:0。图4(b)是相应的出射光谱形状。去离子水作为系统的定标样品,用来调节和衡量系统的稳定性。从图4(a)中water对应实验开始前与结束后的中心波长实时采集的图像,看出水的中心波长没有变化,说明本系统已经稳定;(1)为纯右旋脯氨酸对映体,中心波长的偏移大约有-1nm;(5)为纯左旋脯氨酸对映体,中心波长的偏移大约有+1nm,说明相同浓度这两种溶液旋光度的绝对值相同;(3)对应的对映体比例为2:2,这是外消旋对映体混合物,从图中可以看出(3)与water的中心波长处在同一高度,说明本样品完全消旋,其旋光度为0;(2)的对映体混合比例为1:3,消旋后,混合溶液的旋光度相当于2g/l纯右旋脯氨酸溶液产生的旋光度;同理(4)对应的混合溶液的旋光度相当于2g/l纯左旋脯氨酸溶液产生的旋光度。
从图4(b)可以看出,浓度相同的脯氨酸样品,其比例不同,纯度不同,旋光度也不同,出射光谱的形状有微小的差异,主要体现于波峰的强度变化和中心波长的偏移。从以上实验结果可以看出,每次将不同左右旋比例的脯氨酸溶液加入sc中时,输出光谱的中心波长也随之发生偏移。因此,通过这种实时监测,可以直观地观察混合对映体脯氨酸溶液的手性变化,这在药物合成的实时监测和分析中起着特别重要的作用。
2.3不同浓度对映体混合物中心波长检测
图5为四组不同浓度对映体混合物中心波长移动实时采集数据经平滑消噪后的图像,图(a)至(d)的总浓度分别为4g/l,2g/l,1g/l,0.5g/l。每一组实验采集了五种不同比例的脯氨酸对映体数据,其左右旋比例依次为0:4,1:3,2:2,3:1,4:0。从图中可以看出,除总浓度为0.5g/l的这一组实验波动性较大外,其他三组浓度梯度的实验稳定性较好。随着左右旋比例的变化,中心波长发生了均匀地偏移。
基于上述实验数据,脯氨酸对映体混合溶液中心波长偏移对应浓度的响应图像,如图6所示。从图中可以看出,相同比例对映体混合物中心波长的偏移随浓度变化都是线性的,也就是说,图6中斜率相同的对映体溶液其左右旋比例相同。其中,纯左旋(4:0)和纯右旋(0:4)脯氨酸溶液对应的曲线具有最大的斜率,外消旋混合物(2:2)对应的曲线斜率为0,曲线b(3:1)与d(1:3)有较好的对称性。这些实验结果归因于如下原因:混合对映体出射光谱中心波长的偏移就应与消旋后对映体混合物净浓度成正比。
2.4旋光度和纯度计算
脯氨酸对映体混合溶液的旋光度可以由公式(6)计算。相关参数为:中心波长为830nm,带宽为40nm,β为0.02。各组中心波长的偏移与相应的旋光度、纯度计算结果如表1所示。
对比例1:对比实验
为了对比弱测量的测量精度,利用传统的偏光仪对l-proline进行检测。基于偏光检测原理,移去弱测量系统的qwp,并使两个偏振片完全正交就可构成简易偏光仪。图7是偏光仪出射光强随脯氨酸浓度变化的响应图像。
利用偏光仪检测l-proline的浓度结果中,浓度低于6g/l的溶液的测量结果有较大的误差,难以准确测量对应的旋光度,通过估算偏光仪测脯氨酸的浓度分辨率为2.1×10-5mol/ml。本发明提供的方法,旋光度分辨率能够达到2.4×10-4rad。由此可知,用弱测量方法测量手性分子的旋光度比传统偏光仪精确度至少要高出两个数量级。
本发明的范围不受所述具体实施方案的限制,所述实施方案只欲作为阐明本发明各个方面的单个例子,本发明范围内还包括功能等同的方法和组分。实际上,除了本文所述的内容外,本领域技术人员参照上文的描述和附图可以容易地掌握对本发明的多种改进。所述改进也落入所附权利要求书的范围之内。上文提及的每篇参考文献皆全文列入本文作为参考。
- 还没有人留言评论。精彩留言会获得点赞!