一种通窍鼻炎颗粒溶出度测定法的制作方法
本发明具体涉及一种通窍鼻炎颗粒溶出度测定法。
背景技术:
通窍鼻炎颗粒是由黄芪、防风、炒苍耳子、辛夷、炒白术(部分)等6味药经过加水煎煮、过滤、减压浓缩制成的清膏与部分粉碎的炒白术及全量粉碎的白芷混匀,干燥、粉碎后制成的混悬颗粒。
2015年版《中国药典》第四部通则“0104颗粒剂”中规定,混悬颗粒应进行溶出度检查。而目前还没有关于通窍鼻炎颗粒溶出度检查方法的报道,《中国药典》2015年版一部项下通窍鼻炎片质量测定方法中也未列有溶出度检查方法。
技术实现要素:
为解决上述问题,本发明提供了一种通窍鼻炎颗粒溶出度测定法,其特征在于:它包括如下操作步骤:
1)对照品溶液的制备:取欧前胡素对照品,加甲醇溶解,即得;
2)供试品溶液的制备:取通窍鼻炎颗粒,以十二烷基硫酸钠溶液为溶出介质,恒温,匀速,定时溶出,取溶出液离心,以上清液作为供试品溶液;
3)溶出量测定:分别吸取对照品溶液与供试品溶液,注入液相色谱仪,计算通窍鼻炎颗粒中欧前胡素的溶出量;色谱条件为:色谱柱:c18色谱柱,流动相:甲醇-水(54:46)。
进一步地,步骤1)所述欧前胡素对照品溶液浓度为1.5μg/ml。
进一步地,步骤2)所述通窍鼻炎颗粒与十二烷基硫酸钠溶液的质量体积比为2:500g/ml。
更进一步地,所述十二烷基硫酸钠溶液浓度为0.1%。
进一步地,步骤2)所述恒温是溶出介质温度恒定在37℃±0.5℃;所述匀速是设定搅拌桨转速为50r/min;所述定时是溶出时间为45min;所述溶出液取用量为20ml;所述离心的速度为12000r/min,时间为10min。
进一步地,步骤3)所述分别吸取对照品溶液与供试品溶液体积为20μl。
进一步地,步骤3)所述c18色谱柱为inertsilods-2,4.6×150mm5um或shim-packvp-ods,4.6×150mm5um。
进一步地,步骤3)所述色谱条件的检测波长为248nm,柱温为35℃,流速为1ml/min,理论板数按欧前胡素峰计算应不得低于3000。
进一步地,步骤3)所述通窍鼻炎颗粒中欧前胡素的溶出量限度为70%。
本发明的一种通窍鼻炎颗粒溶出度测定法,构建了溶出度测定的条件,确定了溶出介质、搅拌桨转速、取样时间。完善了通窍鼻炎制剂的检测项目,为通窍鼻炎颗粒的质量判定提供了新方法。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
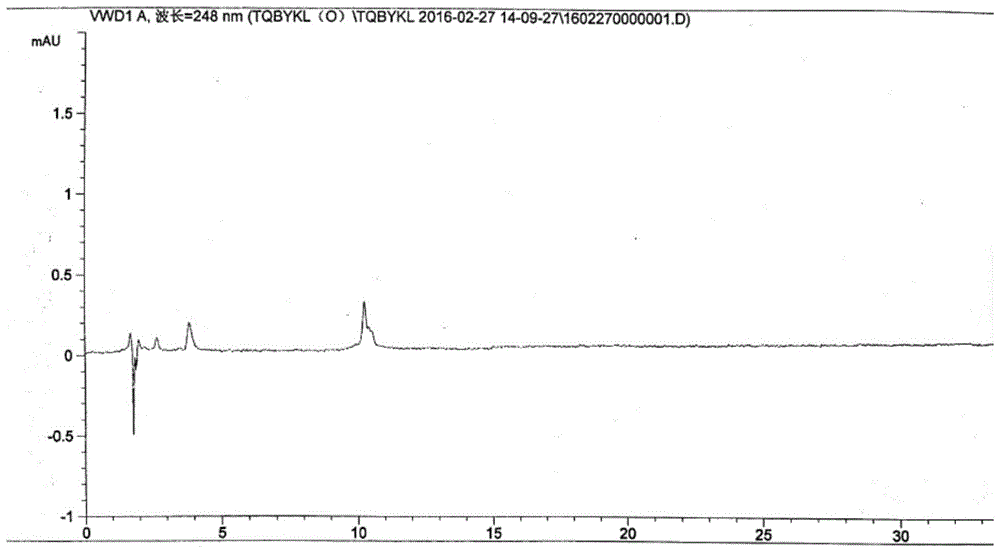
图1空白溶液高效液相色谱图
图2阴性对照溶液高效液相色谱图
图3对照品溶液高效液相色谱图
图4供试品溶液高效液相色谱图
具体实施方式
仪器与试药
仪器:高效液相色谱仪、超声波清洗器、高速离心机、溶出度试验仪、电子天平。
试剂:甲醇(色谱纯),十二烷基硫酸钠(分析纯),超纯水。
对照品:欧前胡素。
样品:通窍鼻炎颗粒。
本发明具体实施方式中使用的试剂、设备、样品均为已知产品,通过购买市售产品获得
实施例1
1)对照品溶液的制备:
取欧前胡素对照品,精密称定,加甲醇制成每1ml含1.5μg的溶液,即得;
2)供试品溶液的制备:
取通窍鼻炎颗粒6袋(每袋重量2g),分别置以0.1%十二烷基硫酸钠溶液500ml为溶出介质的溶出杯中,溶出介质温度恒定在37℃±0.5℃,照《中国药典》2015版四部通则0931溶出度与释放度测定法的第二法(桨法)操作,控制搅拌桨转速为每分钟50转,经45分钟时,取溶液20ml,离心(12000转/分钟,10分钟),取上清液作为供试品溶液;
3)溶出量测定
分别精密吸取对照品溶液与供试品溶液各20ul,注入液相色谱仪,计算通窍鼻炎颗粒中欧前胡素的溶出量。
色谱条件如下:
检测波长:248nm;
色谱柱:以十八烷基硅烷键合硅胶为填充剂的色谱柱,inertsilods-2,4.6×150mm5um或shim-packvp-ods,4.6×150mm5um。流动相:甲醇-水(54:46);
柱温:35℃
流速:1ml/min
通窍鼻炎颗粒中欧前胡素的溶出量限度为70%。
以下通过试验例的方式说明本发明的有益效果:
试验例1本发明方法学考察
1色谱条件
色谱柱:十八烷基硅烷键合硅胶为填充剂的色谱柱,规格4.6×150mm,5μm;
柱温:35℃
流速:1ml/min
检测波长:248nm
流动相:甲醇-水(54:46)
进样量:20μl
2试液的配制:
0.1%十二烷基硫酸钠溶液的配制:称取十二烷基硫酸钠1g,加蒸馏水溶解至1000ml,超声使溶解,摇匀,静置,即得。
3对照品溶液的制备:
3.1线性范围
对照品贮备液(ⅰ):取欧前胡素对照品约5mg,精密称定,置50ml容量瓶中,加甲醇溶解并定容,摇匀,即得0.1mg/ml的欧前胡素对照品贮备液。
对照品贮备液(ⅱ):精密量取上述对照品贮备液(ⅰ)10ml,置100ml容量瓶中,加甲醇稀释并定容,摇匀,即得0.01mg/ml的欧前胡素对照品贮备液。
对照品溶液(ⅰ):精密量取上述对照品贮备液(ⅱ)5ml,置100ml容量瓶中,加甲醇稀释并定容,摇匀,即得0.5μg/ml的对照品溶液。
对照品溶液(ⅱ):精密量取上述对照品贮备液(ⅱ)10ml,置100ml容量瓶中,加甲醇稀释并定容,摇匀,即得1μg/ml的对照品溶液。
对照品溶液(ⅲ):精密量取上述对照品贮备液(ⅱ)15ml,置100ml容量瓶中,加甲醇稀释并定容,摇匀,即得1.5μg/ml的对照品溶液。
对照品溶液(ⅳ):精密量取上述对照品贮备液(ⅱ)20ml,置100ml容量瓶中,加甲醇稀释并定容,摇匀,即得2μg/ml的对照品溶液。
对照品溶液(ⅴ):精密量取上述对照品贮备液(ⅱ)25ml,置100ml容量瓶中,加甲醇稀释并定容,摇匀,即得2.5μg/ml的对照品溶液。
3.2准确度
对照品溶液的制备:取欧前胡素对照品约12.5mg,精密称定,置25ml容量瓶中,加甲醇溶解并定容,摇匀,即得0.5mg/ml的欧前胡素对照品溶液(ⅵ)。
4供试品溶液的制备:照《中国药典》2015版四部通则0931溶出度与释放度测定法的第二法(桨法)设定。分别量取溶出介质500ml置6个溶出杯内,将转速设定为50~75转/分钟。待溶出介质温度恒定在37℃±0.5℃后,取本品6袋(2g/袋),分别投入6个溶出杯内,立即开始计时。至规定取样时间,在桨叶顶端至液面的中点,距溶出杯内壁10mm处,吸取溶出溶液20ml,如需多次取样时,应及时补加等量温度为37℃±0.5℃的溶出介质,立即将取出的溶液高速离心分离10分钟,取其上清液,即为供试品溶液。
5溶出条件的选择
5.1溶出介质的选择
按供试品溶液的制备方法,分别以水和0.1%十二烷基硫酸钠为溶出介质,转速设置为75转/分钟,分别在30min,45min,60min取样,按1项下的色谱条件测定其峰面积,计算溶出量和溶出量的平均值,选择溶出量较高的0.1%十二烷基硫酸钠为溶出介质。
5.2溶出度测定转速和取样时间的选择
转速的选择:按供试品溶液的制备方法,使用5.1项下溶出量较高的0.1%十二烷基硫酸钠为溶出介质,分别设置转速为50转/分钟和75转/分钟,分别在30min,45min,60min取样,按1项下的色谱条件测定其峰面积,计算溶出量和溶出量的平均值,选择溶出量与75转/分钟相似并且转速相对较低的50转/分钟为转速;
取样时间的选择:选择连续两个时间点达到最大溶出量的后者45min为溶出时间。
5.3溶出曲线的制备
按5.1和5.2选择的溶出条件,选取2批通窍鼻炎颗粒(批号:150609、150612),在5min、10min、15min、20min、30min、45min的6个时间点分别取样,按1项下的色谱条件测定其峰面积,计算溶出量和溶出量的平均值,按下式计算相似因子(f2)。
式中rt和tt分别为两制剂在第n个取样点时的平均累计溶出量。
经计算得出:溶出曲线的相似因子(f2)为:83.84,符合相似因子(f2)应≥50的标准。
5.4系统适用性
连续用对照品溶液进样5次,供试品溶液进样1次,记录色谱图、理论板数及对称因子,以5次进样峰面积的相对标准偏差为重复性,供试品溶液色谱图中欧前胡素色谱峰与最近的杂质峰的分离度为分离度。
分离度计算公式:
式中r为分离度
tr1为相邻两峰中后一峰的保留时间
tr2为相邻两峰中前一峰的保留时间
w1与w2分别为相邻两峰的峰宽
对称因子计算公式:
式中t为对称因子
w0.05h为5%峰高处的峰宽
d1为峰顶点至峰前沿之间的距离
理论塔板数计算公式:
式中n为理论塔板数
tr为保留时间
w为峰宽
经计算理论塔板数平均值为4039,rsd(%)为1.6%;欧前胡素色谱峰与相邻色谱峰的分离度为5.97;对称因子平均值为1.14);对照品溶液5次进样峰面积的平均值为158,rsd(%)为0.5%。符合理论板数,按欧前胡素峰计算应≥3000;欧前胡素色谱峰与相邻色谱峰的分离度应≥1.5;0.8≤对称因子≤1.5;重复性,rsd应≤2.0%的标准。
5.5线性
使用紫外检测器检测,欧前胡素对照品的浓度应与峰面积成一次线性关系。以0.01mg/ml的欧前胡素对照品溶液为贮备溶液,分别稀释制成0.5μg/ml、1μg/ml、1.5μg/ml,2μg/ml,2.5μg/ml系列浓度的对照品溶液(对照品溶液ⅰ-ⅴ),作为线性测定溶液。每种浓度的对照品溶液分别进样3次,记录色谱图,以平均峰面积对浓度进行最小二乘法线性回归,得到回归方程,相关系数。回归方程:y=0.01x+0.0046;相关系数:1.0000,符合可接受标准相关系数应≥0.998的标准。
5.6精密度
5.6.1重复性:
取6袋样品,分别称重,按供试品溶液的制备方法制备6份供试品溶液,并按1项下的色谱条件测定其峰面积,计算溶出量和溶出量的相对标准偏差。
5.6.2中间精密度:
由2名操作人员分别对同一批样品进行测定,按供试品溶液的制备方法分别制备6份供试品溶液,并按1项下的色谱条件测定其峰面积,计算溶出量和溶出量的相对标准偏差。
由同一操作人员对同一批样品,按供试品溶液的制备方法制备6份供试品溶液,分别使用两台仪器,按1项下的色谱条件测定其峰面积,计算溶出量和溶出量的相对标准偏差。
由同一操作人员对同一批样品,在不同日期按供试品溶液制备方法分别制备6份供试品溶液,按1项下的色谱条件测定其峰面积,计算溶出量和溶出量的相对标准偏差。
重复性试验rsd(%)0.9%;中间精密度(不同人员)rsd(%)0.9%;中间精密度(不同仪器)rsd(%)1.1%;中间精密度(不同时间)rsd(%)1.2%,符合相对标准偏差应≤2.0%的可接受标准。
5.7专属性
空白溶液:仅含溶出介质的溶液。
对照品溶液:线性范围项下对照品溶液(ⅲ)。
阴性对照溶液:取按通窍鼻炎颗粒配方比例制成的不含白芷药材的颗粒约2g,精密称定,按供试品溶液的制备方法制备,即得阴性对照溶液。
供试品溶液:按供试品溶液的制备方法制备。
分别取空白溶液,阴性对照溶液,对照品溶液,供试品溶液进样1次,记录色谱图,分别见图1、图2、图3、图4。从图中可见空白溶液色谱图、阴性对照溶液色谱图均无干扰;供试品溶液主成分的色谱峰与其他杂质峰完全分离,分离度大于1.5;阴性对照无干扰。
5.8准确度
照《中国药典》2015版四部通则0931溶出度与释放度测定法的第二法(浆法),分别量取溶出介质500ml置6个溶出杯内,将转速设定至50转/分钟。待溶出介质温度稳定在37℃±0.5℃后,分别精密量取对照品溶液(ⅵ)2ml加至6个溶出杯内,立即开始计时,在45min时,在桨叶顶端至液面的中点,距溶出杯内壁10mm处,吸取溶出溶液20ml,立即将取出的溶液高速离心分离10分钟,取其上清液,即得到准确度试验用供试品溶液,按1项下的色谱条件测定其峰面积,计算回收率和回收率的相对标准偏差。
回收率的计算公式:回收率(%)=测得量/加入量×100%
经计算,回收率范围在100.15%~103.17%之间,回收率相对标准偏差rsd1.2%。符合92%≤回收率≤105%,回收率的相对标准偏差应≤2.0%的可接受标准。
5.9耐用性
5.9.1分析方法对流动相比例变化的耐受性
在其他色谱条件不变的前提下,改变流动相的比例,分别为:甲醇-水(50:50),甲醇-水(60:40),不同流动相比例条件下,对照品溶液分别连续进样5次,供试品溶液进样1次,系统适用性符合5.4项下的标准。
5.9.2分析方法对色谱柱变化的耐受性
在其他色谱条件不变的前提下,改变色谱柱的品牌,对照品溶液分别连续进样5次,供试品溶液进样1次,系统适用性符合5.4项下的标准。
5.9.3供试品溶液的稳定性
供试品溶液制备完成后,在室温下密闭放置,在0,2,4,8,12,24h分别进样3次,计算平均峰面积的相对标准偏差。2小时及以后时间点相对于0时间点的相对标准偏差分别为:0,0.1%,0.3%,0.6%,0.7%,符合各考察时间点,供试品溶液的峰面积平均值与0时间点的峰面积平均值比较的相对平均偏差应≤2.0%的可接受标准。
6溶出限度的确定
取10批通窍鼻炎颗粒样品,按照4项下方法分别制备6份供试品溶液,按照1项下色谱条件测定其溶出量,10批通窍鼻炎颗粒溶出量的平均值结果范围为:77.76%~94.09%,从而以此结果范围制定出通窍鼻炎颗粒中欧前胡素合理的溶出量限度标准,即欧前胡素溶出量的限度为按标示装量计算所得每袋含量的70%。
通过方法学考察,证实了本发明的通窍鼻炎颗粒溶出度测定法,重复性强,稳定性好,准确度和精密度高,能用于通窍鼻炎颗粒的质量判定,完善了现行的通窍鼻炎制剂的检测项目。
- 还没有人留言评论。精彩留言会获得点赞!