一种动物体内鬼臼毒素的快速检测方法与流程
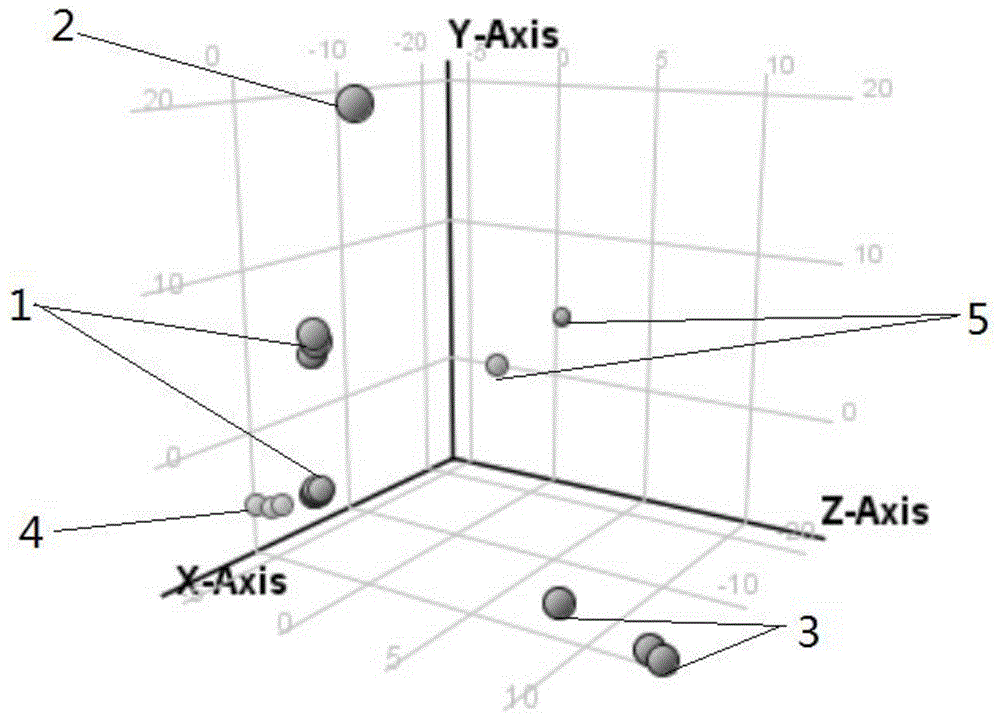
本发明涉及食品毒素的检测方法,特别涉及一种动物体内鬼臼毒素的快速检测方法。技术背景鬼臼毒素是从小蘖科鬼臼属植物---华鬼臼(又称鸡苔素)的根和茎中提取到的木脂类抗肿瘤成份,为白色结晶性粉末,无嗅,有吸湿性。鬼臼生于海拔300~2400米的山坡林下、灌丛中、溪旁阴湿处、竹林下或石灰山常绿林下。分布于湖南、湖北、浙江、江西、安徽、广东、广西、云南、贵州、四川、河南、陕西。使用不当极其容易发生中毒事故,误服可引起严重系统性毒性作用,可发生肾功能衰竭和肝脏中毒(血清乳酸脱氢酶、门冬氨酸氨基转移酶和碱性磷酸增高);神经系统反应发生较迟,持续时间较长,脑中毒可表现触性皮炎,系统性中毒的初起症状有腹疼或胃部疼痛、手脚不灵活觉、恶心、呕吐、腹泻,严重者或日久,可致白细胞及血小板减少,系统中毒的延缓症状有植物神经紊乱,如排尿困难、尿痛、头晕或头轻感(特别从坐位或卧位起立时),心跳加快、呼吸困难、倦睡、麻痹性肠梗阻、周围神经病变、抽搐等。口服鬼臼毒素300mg即可致死。目前,对动物体内鬼臼药物含量检测方法报道的文献较少。所以,开发一种快速、准确、稳定的检测生物样品中鬼臼毒素含量的方法尤为重要。技术实现要素:本发明提供了一种动物体内鬼臼毒素的快速检测方法,该方法可以对鬼臼毒素中毒结果进行更快速准确的检测。为解决上述技术问题,本发明提供了以下的技术方案:一种动物体内鬼臼毒素的快速检测方法,包括以下步骤:(1)取中毒动物体液,配制成待测样液;(2)将所述步骤(1)得到的待测样液用去蛋白剂除杂、净化后,再用高效液相色谱-质谱法测定,即可。进一步地,所述步骤(2)中的高效液相色谱采用3×100mm,1.9μm的c18色谱柱;进样量为1μl;柱温为35℃;5mmol/l乙酸铵-0.1%甲酸为流动相a,乙腈为流动相b进行梯度洗脱,其中流动相a:流动相b的体积比为90~10:10~90。进一步地,所述步骤(2)中的质谱条件:分辨率为70000,毛细管温度为320℃,辅助气温度为350℃;扫描质量范围为60~700amu,扫描模式为全扫描+全碎裂。进一步地,所述步骤(2)中待测样液与去蛋白剂的体积比为1-3:4-6。进一步地,所述步骤(2)中待测样液与去蛋白剂的体积比为2:5。进一步地,所述去蛋白剂为1%~3%的甲酸乙腈溶液。进一步地,所述去蛋白剂为1%的甲酸乙腈溶液。进一步地,所述步骤(1)中,中毒动物体液为尿液、血液和胃内容物中的一种或几种。与现有技术相比,本发明的动物体内鬼臼毒素快速检测方法的有益效果为:该方法简单易行、通过高效液相色谱-质谱法检测,操作方便;结合hrms分析,可快速准确检测出动物体内鬼臼毒素的含量,有助于后期对于治疗药物的筛选和评价。附图说明下面结合附图中的具体实施例对本发明的技术方案做进一步的详细说明,但不构成对本发明的任何限制。图1为鬼臼毒素的主成分分析对比图之一;图2为鬼臼毒素的主成分分析对比图之二;图3为鬼臼毒素的主成分分析对比图之三。其中,1-空白组;2-臼毒鬼素提取液;3-鬼臼毒素提取液-血液;4-鬼臼毒素提取液-胃内容物;5-鬼臼毒素提取液-尿液。具体实施方式实施例1取中毒小鼠尿液2ml配制成待测样液,然后取待测样液1ml加入4ml1%的甲酸乙腈溶液除杂、净化后,再用高效液相色谱-质谱法测定。其中,高效液相色谱采用3×100mm,1.9μm的c18色谱柱;进样量为1μl;柱温为35℃;5mmol/l乙酸铵-0.1%甲酸为流动相a,乙腈为流动相b进行梯度洗脱,其中,0-2min,流动相a与流动相b的体积比为90:10,2-5min,流动相a与流动相b的体积比为10:90,5-5.1min,流动相a与流动相b的体积比为10:90,5.1-7min,流动相a与流动相b的体积比为90:10,第7min,流动相a与流动相b的体积比为90:10;所述质谱条件:分辨率为70000,毛细管温度为320℃,辅助气温度为350℃;扫描质量范围为60~700amu,扫描模式为全扫描+全碎裂。实施例2取中毒小鼠血液3ml,血液置于肝素钠润洗过的离心管中混匀,配制成待测样液,然后取待测样液2ml加入5ml1%的甲酸乙腈溶液除杂、净化后,再用高效液相色谱-质谱法测定。其中,高效液相色谱采用3×100mm,1.9μm的c18色谱柱;进样量为1μl;柱温为35℃;5mmol/l乙酸铵-0.1%甲酸为流动相a,乙腈为流动相b进行梯度洗脱,其中,0-2min,流动相a与流动相b的体积比为90:10,2-5min,流动相a与流动相b的体积比为10:90,5-5.1min,流动相a与流动相b的体积比为10:90,5.1-7min,流动相a与流动相b的体积比为90:10,第7min,流动相a与流动相b的体积比为90:10;所述质谱条件:分辨率为70000,毛细管温度为320℃,辅助气温度为350℃;扫描质量范围为60~700amu,扫描模式为全扫描+全碎裂。实施例3取中毒小鼠胃内容物6ml,于2ml1%的甲酸水溶液中剪碎常温浸泡30min配制成待测样液,然后取待测样液3ml加入6ml1%的甲酸乙腈溶液除杂、净化后,再用高效液相色谱-质谱法测定。其中,高效液相色谱采用3×100mm,1.9μm的c18色谱柱;进样量为1μl;柱温为35℃;5mmol/l乙酸铵-0.1%甲酸为流动相a,乙腈为流动相b进行梯度洗脱,其中,0-2min,流动相a与流动相b的体积比为90:10,2-5min,流动相a与流动相b的体积比为10:90,5-5.1min,流动相a与流动相b的体积比为10:90,5.1-7min,流动相a与流动相b的体积比为90:10,第7min,流动相a与流动相b的体积比为90:10;所述质谱条件:分辨率为70000,毛细管温度为320℃,辅助气温度为350℃;扫描质量范围为60~700amu,扫描模式为全扫描+全碎裂。实验例11.鬼臼药酒的配置称取100g鬼臼,放到酒瓶中,加20°米酒1000ml,泡制1个月,制成鬼臼药酒。2.提取鬼臼药酒中的鬼臼毒素提取液,并检测浓度量取5ml鬼臼药酒于离心管中,加入95ml1%甲酸水溶液稀释,超声提取30分钟,3000rpm离心5分钟。取20ml上清液于50ml离心管中,振荡3分钟,15000rpm离心5分钟。取上清液2.00ml直接上pcx柱(柱子先用3ml甲醇和3ml水活化),再用3ml水和3ml甲醇淋洗固相萃取柱,抽干,用5ml5%氨化甲醇洗脱,收集洗脱液,并用40℃氮吹干,然后用1.00ml0.1%甲酸甲醇定容,过0.22μm有机滤膜,用色谱仪进行检测。色谱条件为安捷伦hph-c18(3×100mm,1.9μm)、进样量1μl,柱温为35℃;质谱条件为:分辨率为70000,毛细管温度为320℃,辅助气温度为350℃;扫描质量范围为60~700amu,扫描模式为全扫描+全碎裂。检测浓度的结果如表1所示,鬼臼药酒中鬼臼毒素的浓度为1664871.6534ng/ml。表1鬼臼毒素提取液浓度检测结果药酒生物碱鬼臼毒素含量(ng/ml)鬼臼药酒1664871.65343.动物模型的制备取10只spf级雌性小鼠并分为两组,第一组:8只(1-8号)灌以上述提取的鬼臼药酒(1.0ml/10g)(1.0ml/10g是指小鼠的体重每10g加1.0ml的提取液);第二组:2只(8-10号)作为空白组,灌以20°米酒(1.0ml/10g)。4.体液的采集和处理方法上述小鼠灌胃后放入代谢笼观察记录动物行为,3h后取血液、胃内容物,收集实验过程中的尿液。(1)尿液:收集代谢笼所得小鼠尿液,取0.4ml加入1%的甲酸乙腈溶液1.0ml混匀,离心上机分析。(2)血液:血液置于肝素钠润洗过的离心管中混匀,离心取血浆0.4ml加入1%的甲酸乙腈溶液1.0ml混匀,离心上机分析。(3)胃内容物:取小鼠胃内容物于1%甲酸水溶液2ml中剪碎常温浸泡30min,离心取上清液0.4ml,加入1%的甲酸乙腈溶液1.0ml混匀,离心上机分析。以上分析条件为:色谱条件:高效液相色谱采用3×100mm,1.9μm的c18色谱柱;进样量为1μl;柱温为35℃;5mmol/l乙酸铵-0.1%甲酸为流动相a,乙腈为流动相b进行梯度洗脱,流动相a和流动相b的体积比见表2;质谱条件:分辨率为70000,毛细管温度为320℃,辅助气温度为350℃;扫描质量范围为60~700amu,扫描模式为全扫描+全碎裂。表2各时间段流动相体积比例5.中毒小鼠体液分析对中毒小鼠体液进行检测分析,条件与上述4中的相同。对小鼠灌胃之后的3h的行为表现以及体液分析结果如表3所示,对小鼠灌胃鬼臼毒素之后均出现呼吸急促的表现,多个小鼠有明显抽搐的表现,个别小鼠有惊厥的表现,通过动物表现和体液中鬼臼毒素浓度的结果显示,所用提取方法、分析方法在对于体液中鬼臼毒素的应用有效。表3动物实验结果6.统计学差异分析通过hrms分别对血液、胃内容物和尿液中鬼臼毒素的主成分分析,结合massprofilerprofessional软件(利用t-testp<0.05和强度倍数变化fc>2过滤)分析,分别得到图1、2、3中的胃内容物、血液以及尿液中的成分分析。经过主成分分析,结合色谱对鬼臼毒素提取液、空白组、鬼臼毒素提取液-体液以及发病后小鼠体内鬼臼毒素的数据能明显分析,所用提取方法、分析方法在对于体液中鬼臼毒素的应用有效。其中胃内容物的差异性均较大,尿液、血液次之,所以应急采样中,优先提供胃内容物进行检验。由以上筛选方法通过动物实验结合hrms模拟测定动物服用鬼臼毒素后,体内胃内容物、血液、尿液中鬼臼毒素的分布情况,可以对动物中毒后体内鬼臼毒素含量进行快速准确的检测。以上所述为本发明的优选实施方式,并不限制于本发明。对本领域技术人员来说,在不脱离本发明原理的前提下做出的若干改进和变型,也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1