一种银/碳纳米管复合材料及其制备方法和应用与流程
本发明属于复合材料技术领域,特别涉及一种银/碳纳米管复合材料及其制备方法和应用。
背景技术:
碳材料由于其优异的性能,例如优异的化学稳定性,大的表面积,高的导电性而成为有吸引力的催化材料。通过贵金属纳米颗粒(例如,ag,au,pt,pd等)改性的碳材料可以很大程度提高催化性能。在这些贵金属纳米粒子中,银纳米粒子(agnps)因其高催化活性,价格便宜,导热性和导电性的优异性能而受到广泛关注。高度分散的银纳米颗粒可以大大提高材料的催化性能。然而,银纳米颗粒易团聚,导致电化学活性下降。为了改善这个问题,首先将银纳米粒子均匀地固定在碳材料前驱体上,然后通过碳材料前驱体的碳化直接获得银纳米颗粒改性碳材料,碳材料前驱体包括聚苯胺(pani),聚吡咯(ppy),三聚氰胺,聚多巴胺(pda),聚乙烯吡咯烷酮(pvp)和聚丙烯腈(pan)等。
在这些前体中,聚多巴胺(pda)已广泛研究用于生物传感器,催化剂,吸附剂,抗菌剂,电容器等各种应用。多巴胺(da)可以在碱性溶液中自聚合并表现出优异的粘附性能,其可以自发地附着在各种固体材料的表面上。此外,由于聚多巴胺自身的还原能力,可以和多种金属离子作用而获得金属纳米颗粒。同时聚多巴胺由于含有大量的胺官能团和邻苯二酚,聚多巴胺可以很容易地碳化成氮掺杂的石墨化碳,这可以提高电子传导性和催化活性。负载银、金等的聚多巴胺多有报道,但金属颗粒的负载和聚多巴胺的厚度目前仅采用不同的生长时间及浓度调节,常常导致生长周期过长引起的金属纳米颗粒的团聚(one-stephydrothermalgreensynthesisofsilvernanoparticle-carbonnanotubereduced-grapheneoxidecompositeanditsapplicationashydrogenperoxidesensor,sens.actuatorb-chem.,208(2015)389-398.)。
技术实现要素:
本发明所要解决的技术问题是提供一种银/碳纳米管复合材料及其制备方法和应用,不仅克服了银纳米颗粒与碳材料结合不强和分布不均匀等缺陷,还可得到管壁厚度可调的银/碳纳米管复合材料,同时由聚多巴胺碳化可得到高比表面积、多孔的、富氮的碳纳米管。
本发明的一种银/碳纳米管复合材料,将聚多巴胺和银纳米颗粒包覆在聚苯乙烯纳米纤维表面,采用层层自主装调节银/聚多巴胺厚度,利用高温碳化直接去除聚苯乙烯纤维模板制得。
所述银/碳纳米管复合材料呈管状结构,表面富氮多孔,管壁厚度可控。
所述磺化聚苯乙烯纳米纤维的直径为100-800nm,优选200-300nm。
所述银纳米颗粒的粒径为4-8nm。
所述银/碳纳米管复合材料管壁厚度为30-100nm。
所述聚多巴胺和银纳米颗粒的包覆层数为≥1层,优选1-3层,最优选2层。
本发明还提供了上述银/碳纳米管复合材料的制备方法,包括:
(1)将磺化聚苯乙烯纳米纤维浸入多巴胺溶液中反应,经水洗得到聚多巴胺修饰的聚苯乙烯纳米纤维;
(2)将步骤(1)制得的聚多巴胺修饰的聚苯乙烯纤维浸渍于硝酸银溶液中,然后水洗、真空干燥,得到银纳米颗粒/聚多巴胺修饰的聚苯乙烯纳米纤维;
(3)重复步骤(1)和(2),得到不同层数的银纳米颗粒/聚多巴胺修饰的聚苯乙烯纳米纤维,碳化处理,得到银/碳纳米管复合材料。
所述步骤(1)中多巴胺溶液为含有0.5-10mg/ml,优选2-3mg/ml的多巴胺的10mmtris-hcl,ph=8.5的水溶液。
所述步骤(1)中反应的工艺参数为:反应温度为20-30℃,反应时间为1-24h。
所述步骤(2)中硝酸银溶液的浓度为0.1-50mg/ml。
所述步骤(2)中浸渍的时间为0.5-20h。
所述步骤(3)中碳化处理的工艺条件为:在氮气氛围中,从室温以升温速率2-10℃/min,优选5℃/min,升温至500-1200℃,优选800℃,保持时间30-120min,优选60min。
本发明还进一步提供了上述银/碳纳米管复合材料在过氧化氢电化学传感器或燃料电池催化剂领域中的应用。
本发明将磺化后的聚苯乙烯纳米纤维浸泡于多巴胺溶液中,聚多巴胺自聚包裹在聚苯乙烯纳米纤维表面;后浸入硝酸银溶液中,利用聚多巴胺自身的还原性,在其表面原位还原银纳米颗粒;反复上诉步骤,多层膜在惰性气体下高温碳化,层数不同,得到不同管壁厚度的银/碳纳米管复合材料。银纳米颗粒分布均匀,制备出的银/碳纳米管复合材料富氮多孔,管壁厚度可控,该复合材料是制备活性电催化剂用于过氧化氢电化学传感器,燃料电池催化剂领域表现出良好的前途。
有益效果
(1)本发明设计思路巧妙,将聚多巴胺和银纳米颗粒包覆在聚苯乙烯纳米纤维表面,利用高温碳化直接去除聚苯乙烯纤维模板,得到银/碳纳米管复合材料。本发明制备方法简单易行,反应过程温和、环保,易于操作,是一种绿色化学制备方法。
(2)本发明制备的银/碳纳米管,表面银纳米颗粒分布均匀,具有高比表面积、多孔的、富含氮的管状结构;本发明可通过控制聚多巴胺和银纳米颗粒层层自主装的层数来调控银/碳纳米管的管壁厚度。
(3)本发明制备的银/碳纳米管复合材料用于过氧化氢电化学传感器和燃料电池催化剂,具有高的催化性能。
附图说明
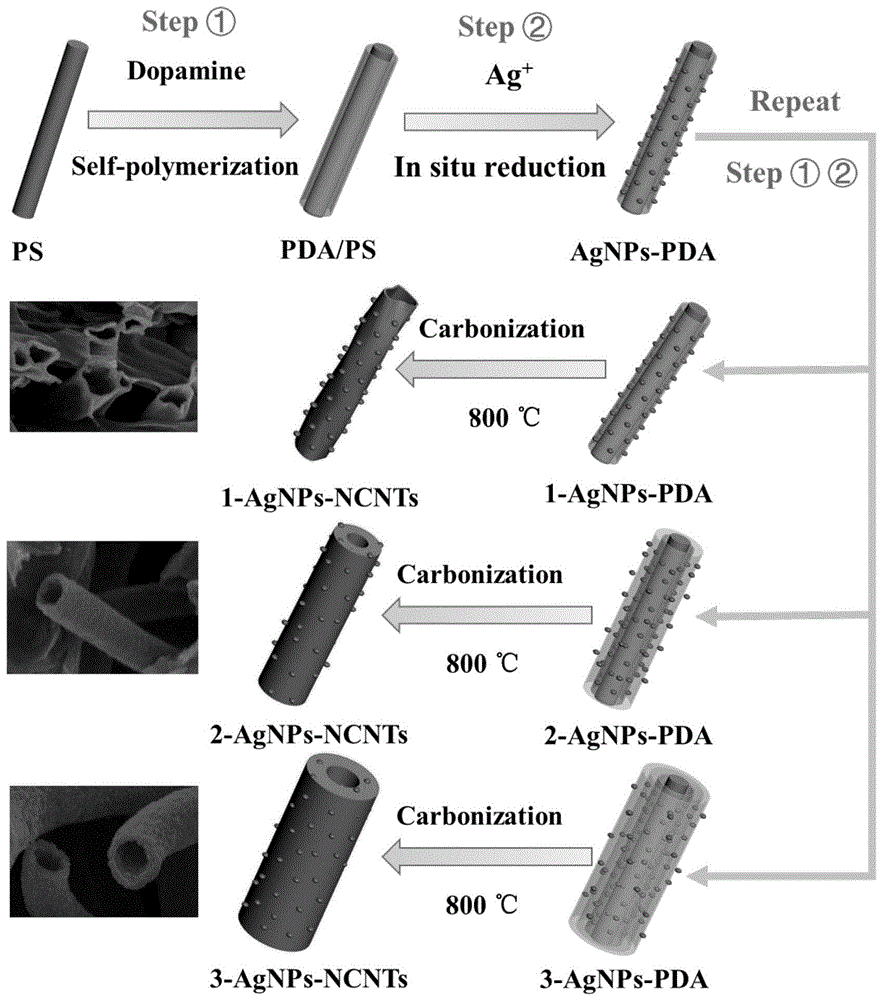
图1是本发明实施例1-3的银/碳纳米管复合材料的制备过程示意图;
图2是本发明实施例1中的聚苯乙烯纳米纤维(a),银/聚多巴胺修饰的聚苯乙烯纳米纤维(b),银/碳纳米管复合纳米材料(c)的扫描电镜图;
图3是本发明实施例1-3制备的不同层数的银/碳纳米管复合材料和对比例1制备的碳纳米管材料的平面和截面扫描电镜图;
图4是本发明实施例2制备的银/碳纳米管复合材料的透射电镜图;
图5是本发明对比例2制备的不同反应时间的银/碳纳米管复合材料扫描电镜图;
图6是本发明实施例1-3制备的不同层数的银/碳纳米管复合材料和对比例1制备的碳纳米管材料的xrd谱图;
图7是本发明实施例2制备的银/碳纳米管复合材料的氮气吸脱附图;
图8是本发明实施例2制备的银/碳纳米管复合材料的n1s的xps图;
图9是本发明实施例2制备的银/碳纳米管复合材料修饰电极在含有不同浓度过氧化氢的磷酸缓冲液中的循环伏安图(a)和浓度与电流的线性图(b);
图10是本发明对比例1制备的裸电极和碳纳米管修饰电极,实施例2制备的银/碳纳米管复合材料修饰电极在氧饱和的磷酸缓冲液中的循环伏安图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
本发明实施例中采用多巴胺(j&kscientificltd);使用μ-autolab-iii电化学工作站在常规三电极系统中进行电化学测试。
实施例1
(1)将磺化后的ps纳米纤维浸入含有2mg/mlda的10mmtris-hcl,ph=8.5的水溶液中反应20h。反应后,为了除去未粘附的pda,用超纯水彻底洗涤纳米纤维数次,得到pda修饰的ps纳米纤维。
(2)将步骤(1)得到的pda修饰的ps纳米纤维浸入10mg/ml的agno3溶液中反应1.5h,用超纯水洗涤数次后放入真空干燥箱在50℃下干燥8h,得到ag/pda修饰的ps纳米纤维。
(3)将步骤(2)得到的样品转移至管式炉中,在氮气的气氛下,以5℃/min的升温速度升至800℃,保留1h,进行碳化,得到ag/碳纳米管复合材料,记作1-agnps-ncnts。
本实施例中ps纳米纤维的扫描电镜图如图2a所示,ag/pda修饰的ps纳米纤维的扫描电镜图如图2b所示,ag/碳纳米管复合材料的扫描电镜图如图2c所示,其平面和截面的扫描电镜图分别如图3b和b'所示,可知ag/碳纳米管复合材料的尺寸均匀,有一定程度的坍塌。
本实施例制得的ag/碳纳米管复合材料的xrd谱图如图6所示,其中x-射线衍射曲线中的2θ=38.2°、44.2°、64.4°、77.4°和81.6°处的衍射峰与银的标准衍射峰一致(jcpds,编号04-0783),表明银离子通过pda原位还原成银纳米颗粒。
实施例2
(1)将磺化后的ps纳米纤维浸入含有2mg/mlda的10mmtris-hcl,ph=8.5的水溶液中反应20h。反应后,为了除去未粘附的pda,用超纯水彻底洗涤纳米纤维数次,得到pda修饰的ps纳米纤维。
(2)将步骤(1)得到的pda修饰的ps纳米纤维浸入10mg/ml的agno3溶液中反应1.5h,用超纯水洗涤数次后,重复浸入相同条件的多巴胺溶液和硝酸银溶液中。洗涤后放入真空干燥箱在50℃下干燥8h,得到ag/pda修饰的ps纳米纤维。
(3)将步骤(2)得到的样品转移至管式炉中,在氮气的气氛下,以5℃/min的升温速度升至800℃,保留1h,进行碳化,得到ag/碳纳米管复合材料,记作2-agnps-ncnts。
本实施例制得的ag/碳纳米管复合材料的扫描电镜图如图3c(平面)和c'(截面)所示,可知ag/碳纳米管尺寸均匀,成均匀管状结构,ag/碳纳米管直径在200-300nm,管壁厚度在40-60nm。
本实施例制得的ag/碳纳米管复合材料的透射电镜图,如图4所示,表明银纳米颗粒分布均匀,粒径大小约为6nm。
本实施例制得的ag/碳纳米管复合材料的xrd谱图如图6所示,其中x-射线衍射曲线中的2θ=38.2°、44.2°、64.4°、77.4°和81.6°处的衍射峰与银的标准衍射峰一致(jcpds,编号04-0783),表明银离子通过pda原位还原成银纳米颗粒。
本实施例制得的ag/碳纳米管复合材料的氮气吸脱附和比表面积大小如图7所示,表明制得复合材料比表面积为245.1。
本实施例制得的ag/碳纳米管复合材料的n1s的xps谱图如图8所示,n主要以石墨化n和吡啶n的形式存在。
将本实施例制得的ag/碳纳米管分散在无水乙醇中(质量浓度为0.1mg/ml)并用细胞破碎仪进行冰水浴超声30分钟,然后加入nafion,使nafion分散浓度为0.01%,继续超声10分钟使之分散均匀。用微量进样器吸取5μl滴涂在玻碳电极表面,然后自然晾干成膜,得到ag/碳纳米管复合材料修饰的玻碳电极。
以本实施例制得的ag/碳纳米管修饰的玻碳电极作为工作电极,铂丝为对电极,饱和甘汞电极为参比电极,三电极插入10ml磷酸缓冲液(pbs,ph=8,0.1mol/l)中,将不同浓度的过氧化氢加入缓冲液中,接通电化学工作站,进行循环伏安扫描,循环伏安图(a)和浓度与电流的线性图(b)如图9所示,可知随着浓度的增加,相应电流依次加大,在1μm-5mm浓度范围内成良好的线性关系。
实施例3
(1)将磺化后的ps纳米纤维浸入含有2mg/mlda的10mmtris-hcl,ph=8.5的水溶液中反应20h。反应后,为了除去未粘附的pda,用超纯水彻底洗涤纳米纤维数次,得到pda修饰的ps纳米纤维。
(2)将步骤(1)得到的pda修饰的ps纳米纤维浸入10mg/ml的agno3溶液中反应1.5h,用超纯水洗涤数次后,重复两次浸入相同条件的多巴胺溶液和硝酸银溶液中。洗涤后放入真空干燥箱在50℃下干燥8h,得到ag/pda修饰的ps纳米纤维。
(3)将步骤(2)得到的样品转移至管式炉中,在氮气的气氛下,以5℃/min的升温速度升至800℃,保留1h,进行碳化,得到ag/碳纳米管复合材料,记作3-agnps-ncnts。
本实施例制得的ag/碳纳米管复合材料的扫描电镜图如图3d(平面)和d'(截面)所示,可知ag/碳纳米管尺寸均匀,成均匀管状结构,纳米管直径和管壁变厚。
本实施例制得的ag/碳纳米管复合材料的xrd谱图如图6所示,其中x-射线衍射曲线中的2θ=38.2°、44.2°、64.4°、77.4°和81.6°处的衍射峰与银的标准衍射峰一致(jcpds,编号04-0783),表明银离子通过pda原位还原成银纳米颗粒。
对比例1
本发明的对照组,不掺杂银纳米颗粒的碳纳米管的制备,包括:
(1)将磺化后的ps纳米纤维浸入含有2mg/mlda的10mmtris-hcl,ph=8.5的水溶液中反应20h。反应后,为了除去未粘附的pda,用超纯水彻底洗涤纳米纤维数次,放入真空干燥箱在50℃下干燥8h,得到pda修饰的ps纳米纤维。
(2)将步骤(1)制得的pda修饰的ps纳米纤维转移至管式炉中在氮气的气氛下以5℃/min的升温速度升至800℃,保留1h,进行碳化,得到碳纳米管,记作ncnts。
本对比例制得的碳化后纳米管的扫描电镜图如图3a(平面)和a'(截面)所示,可知碳纳米管尺寸均匀,有一定程度的坍塌。本对比例制得的碳纳米管的xrd谱图如图6所示,其中x-射线衍射曲线不含有银的衍射峰。
将本对比例制得的碳纳米管分散在无水乙醇中(质量浓度为0.1mg/ml)并用细胞破碎仪进行冰水浴超声30分钟,然后加入nafion,使nafion分散浓度为0.01%,继续超声10分钟使之分散均匀。用微量进样器吸取5μl滴涂在玻碳电极表面,然后自然晾干成膜,得到碳纳米管修饰的玻碳电极。
分别将裸电极baregce、本对比例制得的碳纳米管修饰的玻碳电极ncnts/gce、实施例2制得的ag/碳纳米管修饰的玻碳电极agnps-ncnts/gce作为工作电极,铂丝为对电极,饱和甘汞电极为参比电极,三电极插入10ml磷酸缓冲液(pbs,ph=8,0.1mol/l)中,在氧饱和条件下进行循环伏安扫描,结果如图10所示,显然可知:裸电极对氧还原催化效果很小;碳纳米管修饰电极对氧还原有一定的催化效果;ag/碳纳米管修饰电极对氧还原有很好的催化效果。
对比例2
本发明的对照组,不同反应时间的ag/碳纳米管复合材料的制备,包括:
(1)将磺化后的ps纳米纤维浸入含有2mg/mlda的10mmtris-hcl,ph=8.5的水溶液中反应40h。反应后,为了除去未粘附的pda,用超纯水彻底洗涤纳米纤维数次,得到pda修饰的ps纳米纤维。
(2)将步骤(1)制得的pda修饰的ps纳米纤维浸入10mg/ml的agno3溶液中反应4.5h,用超纯水洗涤数次后放入真空干燥箱在50℃下干燥8h,得到ag/pda修饰的ps纳米纤维。
(3)将步骤(2)制得的样品转移至管式炉中,在氮气的气氛下,以5℃/min的升温速度升至800℃,保留1h,进行碳化,得到ag/碳纳米管复合材料。
本对比例制得的ag/碳纳米管复合材料的扫描电镜图,如图5所示,可知ag/碳纳米管团聚严重,银纳米颗粒尺寸较大。
- 还没有人留言评论。精彩留言会获得点赞!