一种中药或中药组合物制剂的质量检测方法与鉴别应用与流程
本发明是关于一种中药或中药组合物制剂的质量检测方法与鉴别应用,具体地说,是关于一种高效液相色谱法检测中药组合物制剂的方法,以及其用于含天然牛黄、牛黄、天然麝香、麝香、黄芩、黄连、栀子、郁金的中药或中药组合物制剂质量检测(包括鉴别)等应用,属于中医药技术领域。
背景技术:
中药具有多化学成分、多来源同时入药的特性,导致了中药药物的质量控制的复杂性;中药复方制剂,由多味中药药物组合而成,不仅增加了中药成分复杂的分析难度,更使得在中药复方制剂成品中对各原料中药质量的控制更加困难。
中药特征图谱是一种综合的、可量化的质量检测方法,它是建立在中药化学成分系统研究的基础上,主要用于中药材以及中药制剂成品的定性、定量质量分析。建立合适的中药特征图谱,可通过对中药制剂特征性色谱峰的确定,以及多指标性成分的含量测定分析,对中药成品制剂进行质量控制,并通过中药成品制剂的质量表征结果,鉴别其中药制剂所用原料药材的真伪、优劣等质量特点。因此,中药特征图谱的研究和建立,对于保障中药质量、促进中药现代化具有重要意义。
技术实现要素:
本发明的一个目的在于提供一种中药或中药组合物制剂的质量检测方法。
本发明的另一目的在于提供一种鉴别中药或中药组合物制剂中是否含有天然牛黄、牛黄、天然麝香、麝香、黄芩、黄连、栀子、郁金等中药成分的方法。
为实现上述目的,一方面,本发明提供了一种中药或中药组合物制剂的质量检测方法,该方法包括:
将待测中药或中药组合物制剂用溶剂提取后制备成供试品溶液;
对供试品溶液进行高效液相色谱测定,以分析中药或中药组合物制剂的质量;其中,以乙腈(a)-酸水溶液(b)为流动相,采用梯度洗脱,检测波长190-500nm。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法,特别适用于检测的中药或中药组合物制剂中含有以下至少一种中药成分:天然牛黄、牛黄、天然麝香、麝香、黄芩、黄连、栀子、郁金。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,所述流动相中的酸水溶液为体积浓度0.2%-1%的磷酸水溶液。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,进一步,所述检测波长为240nm、262nm、274nm、330nm、360nm、440nm。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,所述高效液相色谱测定过程中,柱温为30~50℃。具体地,本发明中可采用watersacquityhclass超高效液相色谱仪。优选地,本发明中采用的色谱柱为waterscortecsshieldrp18。色谱柱的规格优选可以为2.1mm×100mm,1.6μm。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,所述高效液相色谱测定过程中,梯度洗脱程序如下:
0到5min时,流动相a体积比为5±5%;
5到6min时,流动相a体积比由5±5%上升到10±5%;
6到24min时,流动相a体积比为10±5%上升到12±5%;
24到30min时,流动相a体积比由12±5%上升到17±5%;
30到35min时,流动相a体积比由17±5%上升到20±5%;
35到40min时,流动相a体积比由20±5%上升到32±5%;
40到44min时,流动相a体积比由32±5%上升到38±5%;
44到47min时,流动相a体积比由38±5%上升到45±5%;
47到50min时,流动相a体积比由45±5%上升到52±5%;
50到57min时,流动相a体积比由52±5%上升到92±5%;
57到61min时,流动相a体积比为92±5%;
61到62min时,流动相a体积比由92±5%下降到5±5%。
在本发明的一些优选具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,梯度洗脱程序如下:
0到5min时,流动相a体积比为8%;
5到6min时,流动相a体积比由8%上升到13%;
6到24min时,流动相a体积比为13%上升到15%;
24到30min时,流动相a体积比由15%上升到20%;
30到35min时,流动相a体积比由20%上升到23%;
35到40min时,流动相a体积比由23%上升到35%;
40到44min时,流动相a体积比由35%上升到40%;
44到47min时,流动相a体积比由40%上升到48%;
47到50min时,流动相a体积比由48%上升到55%;
50到57min时,流动相a体积比由55%上升到95%;
57到61min时,流动相a体积比为95%;
61到62min时,流动相a体积比由95%下降到8%%。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,提取过程中的溶剂为甲醇、乙醇、水、正丁醇、三氯甲烷中任意一种或多种组成的混合溶剂,或上述溶剂与酸水溶液组成的溶剂;所述酸水溶液为甲酸、冰醋酸中的任意一种。优选地,提取过程中的溶剂为体积比50-100:50-0的乙醇:水,或体积比50-100:50-0的甲醇:水。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,待测中药或中药组合物制剂的溶剂提取法可以为超声提取、冷浸提取或回流提取。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法,是用于检测所述中药或中药组合物制剂中是否含有以下至少一种中药成分:天然牛黄、牛黄、天然麝香、麝香、黄芩、黄连、栀子、郁金。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,分析高效液相色谱的图谱中,以盐酸小檗碱色谱峰出峰时间为参照(可根据需要使用盐酸小檗碱对照品),是否出现以下特征峰,以检测所述中药组合物制剂中是否含有对应的中药成分(各相对保留时间允许有±5%、优选±3%、更优选±2%误差):
1号峰,相对保留时间为0.150;240nm下检测;栀子-京尼平龙胆双糖苷;
2号峰,相对保留时间为0.221;240nm下检测;栀子苷;
3号峰,相对保留时间为0.239;330nm下检测;黄连-酚类;
4号峰,相对保留时间为0.290;330nm下检测;栀子-酚类
5号峰,相对保留时间为0.418;330nm下检测;黄连-酚类;
6号峰,相对保留时间为0.446;262nm下检测;天然麝香特征峰;
7号峰,相对保留时间为0.468;330nm下检测;黄连-酚类;
8号峰,相对保留时间为0.544;274nm下检测;黄连-生物碱;
9号峰,相对保留时间为0.579;274nm下检测;黄芩-黄酮;
10号峰,相对保留时间为0.651;274nm下检测;黄芩-黄酮;
11号峰,相对保留时间为0.674;274nm下检测;黄连-盐酸黄连碱;
12号峰,相对保留时间为0.712;274nm下检测;黄连-表小檗碱;
13号峰,相对保留时间为0.759;274nm下检测;黄连-生物碱;
14号峰,相对保留时间为0.794;274nm下检测;黄连-生物碱;
15号峰,相对保留时间为1.000;274nm下检测;黄连-盐酸小檗碱;
16号峰,相对保留时间为1.058;274nm下检测;黄连-盐酸巴马汀;
17号峰,相对保留时间为1.339;240nm下检测;栀子-酚类;
18号峰,相对保留时间为1.378;274nm下检测;黄芩-黄芩苷;
19号峰,相对保留时间为1.524;440nm下检测;栀子-西红花苷1;
20号峰,相对保留时间为1.542;274nm下检测;黄芩-黄酮;
21号峰,相对保留时间为1.624;440nm下检测;栀子-西红花苷2;
22号峰,相对保留时间为1.680;274nm下检测;黄芩-黄酮;
23号峰,相对保留时间为1.682;274nm下检测;黄芩-汉黄芩苷;
24号峰,相对保留时间为1.821;440nm下检测;栀子-二萜类;
25号峰,相对保留时间为1.844;274nm下检测;黄芩-黄芩素;
26号峰,相对保留时间为1.897;440nm下检测;栀子-二萜类;
27号峰,相对保留时间为1.944;440nm下检测;栀子-二萜类;
28号峰,相对保留时间为2.029;274nm下检测;黄芩-汉黄芩素;
29号峰,相对保留时间为2.078;274nm下检测;黄芩-黄酮;
30号峰,相对保留时间为2.228;440nm下检测;郁金-姜黄素;
31号峰,相对保留时间为2.248;440nm下检测;郁金-去甲氧基姜黄素;
32号峰,相对保留时间为2.330;240nm下检测;郁金特征峰;
33号峰,相对保留时间为2.352;360nm下检测;天然牛黄特征峰;
34号峰,相对保留时间为2.398;240nm下检测;郁金特征峰;
35号峰,相对保留时间为2.565;440nm下检测;牛黄特征峰。
在本发明的一些具体实施方案中,本发明的中药或中药组合物制剂的质量检测方法,是用于检测所述中药或中药组合物制剂中是否含有牛黄和/或麝香;
其中,分析高效液相色谱的图谱中,以盐酸小檗碱色谱峰出峰时间为参照,是否出现以下特征峰,以检测所述中药组合物制剂中是否含有对应的中药成分:
6号峰,相对保留时间为0.446;262nm下检测;天然麝香特征峰;
33号峰,相对保留时间为2.352;360nm下检测;天然牛黄特征峰;
35号峰,相对保留时间为2.565;440nm下检测;牛黄特征峰。
在本发明的一些具体实施方案中,本发明的中药或中药组合物制剂的质量检测方法,其中:
同时出现色谱峰1、2、4、17、19、21、24、26、27号色谱峰中的两个或更多个峰,则样品中含有栀子药材;优选地,色谱峰相对峰面积值越大,栀子药材等级质量越好;
同时出现色谱峰3、5、7、8、11、12、13、14、15、16号色谱峰中的两个或更多个峰,则样品中含有黄连药材;优选地,以15号峰的面积为1,11、12、16号色谱峰中的一个或多个峰的相对峰面积值分别大于0.29、0.145、0.2727,判断黄连药材基源为味连;
同时出现色谱峰9、10、18、20、22、23、25、28、29号色谱峰中的两个或更多个峰,则样品中含有黄芩药材;
同时出现色谱峰30、32、34号色谱峰中的两个或三个峰,则样品中含有郁金药材,且药材基源为黄丝郁金;
样品中含有6号色谱峰,则说明样品中含有天然麝香药材;
样品中含有33号色谱峰,则说明样品中含有天然牛黄药材。
本发明的方法,所测定的特征峰,通过各单味药味的缺阴性实验验证,特征峰归属明确,且无阴性干扰,可用于含天然牛黄、牛黄、天然麝香、麝香、黄芩、黄连、栀子、郁金的中药或中药组合物制剂质量检测,通过特征峰的检验可用于天然麝香、天然牛黄及其中药组合物制剂的真伪鉴别。
根据本发明的具体实施方案,本发明的中药或中药组合物制剂的质量检测方法中,所述的中药或中药组合物制剂可以是丸剂、颗粒剂、散剂、片剂、胶囊剂、口服液等。
在本发明的一些具体实施方案中,对含有多味药的中药组合物制剂进行了检测。所述的中药组合物制剂原料药组成为:牛黄50-200重量份、水牛角100-300重量份、麝香(或人工麝香)10-50重量份、珍珠20-100重量份、朱砂50-200重量份、雄黄50-200重量份、黄连50-200重量份、黄芩50-200重量份、栀子50-200重量份、郁金50-200重量份、冰片10-50重量份。这些原料药可制成中药组合物丸剂,具体制备方法为:按比例取原料药,珍珠水飞或粉碎成极细粉;朱砂、雄黄分别水飞成极细粉;黄连、黄芩、栀子、郁金粉碎成细粉;将牛黄、水牛角、麝香(或人工麝香)、冰片研细,与上述粉末配研,过筛,混匀,加适量炼蜜制成大蜜丸,或包金衣,即得。所述中药组合物丸剂中同时含有动物药、植物药和矿物药,化学成分组成复杂,并且其原料中涉及珍贵天然药物,市场价格及质量差距较大。现有技术中尚未见有效可行的质量控制方法,可对其成药制剂进行原料药材质量优劣、真伪的鉴定。而利用本发明的方法,可有效进行质量检测。
综上所述,本发明提供了一种中药或中药组合物制剂的质量检测方法,可用于含天然牛黄、牛黄、天然麝香、麝香、黄芩、黄连、栀子、郁金的中药或中药组合物制剂的质量检测,通过特征峰的检验可用于天然麝香、天然牛黄及其中药组合物制剂的真伪鉴别。
附图说明
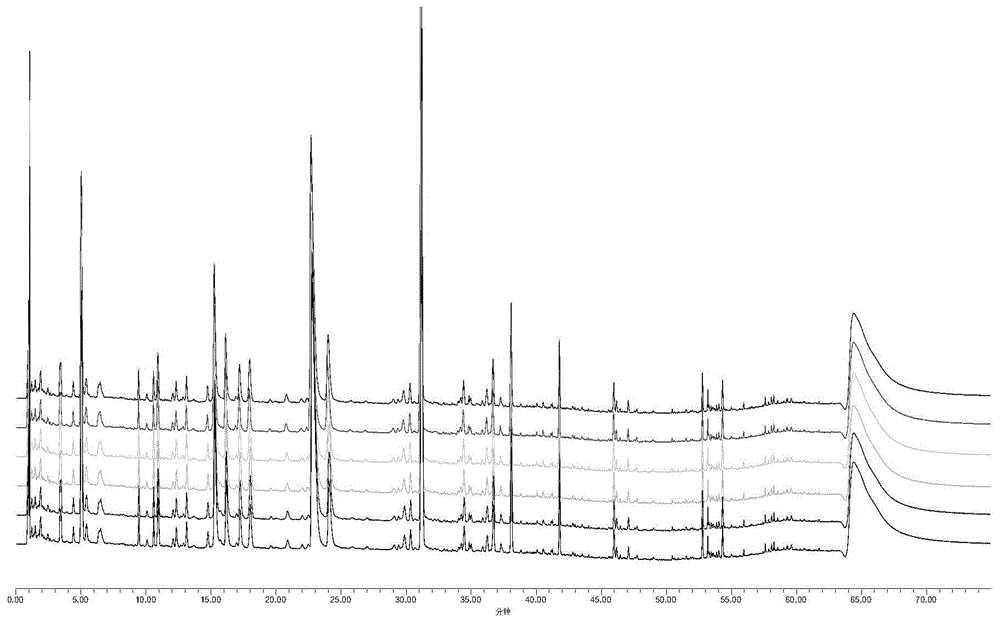
图1为检测波长240nm下anw特征图谱色谱图-精密度。
图2为检测波长262nm下anw特征图谱色谱图-精密度。
图3为检测波长274nm下anw特征图谱色谱图-精密度。
图4为检测波长330nm下anw特征图谱色谱图-精密度。
图5为检测波长360nm下anw特征图谱色谱图-精密度。
图6为检测波长440nm下anw特征图谱色谱图-精密度。
图7为典型的检测波长240nm下anw特征图谱色谱图。
图8为典型的检测波长262nm下anw特征图谱色谱图。
图9为典型的检测波长274nm下anw特征图谱色谱图。
图10为典型的检测波长330nm下anw特征图谱色谱图。
图11为典型的检测波长360nm下anw特征图谱色谱图。
图12为典型的检测波长440nm下anw特征图谱色谱图。
图13为检测波长240nm下anw特征图谱色谱图-稳定性。
图14为检测波长262nm下anw特征图谱色谱图-稳定性。
图15为检测波长274nm下anw特征图谱色谱图-稳定性。
图16为检测波长330nm下anw特征图谱色谱图-稳定性。
图17为检测波长360nm下anw特征图谱色谱图-稳定性。
图18为检测波长440nm下anw特征图谱色谱图-稳定性。
图19为检测波长240nm下anw特征图谱色谱图-重复性。
图20为检测波长262nm下anw特征图谱色谱图-重复性。
图21为检测波长274nm下anw特征图谱色谱图-重复性。
图22为检测波长330nm下anw特征图谱色谱图-重复性。
图23为检测波长360nm下anw特征图谱色谱图-重复性。
图24为检测波长440nm下anw特征图谱色谱图-重复性。
图25为检测波长为240nm下,anw中十一味原料药材特征图谱对比图。
图26为检测波长为274nm下,anw中十一味原料药材特征图谱对比图。
图27为检测波长为330nm下,anw中十一味原料药材特征图谱对比图。
图28为检测波长为440nm下,anw中十一味原料药材特征图谱对比图。
具体实施方式
以下通过具体实施例详细说明本发明技术方案的实施和所具有的有益效果,但不能认定为对本发明的可实施范围的任何限定。各实施例中未详细说明的方法和操作条件,按照所述领域的常规技术或是按照仪器制造商建议的操作进行。
实施例1
1仪器与试药
1.1仪器
watersacquityhclass超高效液相色谱仪(包括四元泵,2996dad检测器,柱温箱,自动进样器,empower工作站);超声波清洗器(kq-250de型数控超声波清洗机,昆山超声仪器有限公司);mettlerml204型电子分析天平(瑞士梅特勒-托利集团);mettlerxp205型电子分析天平(瑞士梅特勒-托利集团),0.22μm微孔滤膜(天津津腾实验设备有限公司)。
1.2试药
乙腈(默克公司,色谱纯),无水甲酸(分析纯,天津光复精细化工研究所),超纯水(milli-q超纯水仪)。对照品:盐酸小檗碱(含量86.8%,批号110713-201613;),盐酸巴马汀(含量86.8%,批号110732-201611)、盐酸黄连碱(含量95.1%,批号:112026-201601)、栀子苷(含量97.6%,批号:110749-201718)、黄芩苷、汉黄芩苷(批号:112002-201702,纯度:98.5%)、黄芩素(批号:111595-201306,纯度:97.8%)、汉黄芩素(批号:111514-201605,纯度:100%)、姜黄素(含量98.9%,批号:110823-201405)均购自中国食品药品检定研究院,表小檗碱(含量98%,批号w06m8z30708)、京尼平龙胆双糖苷(批号:zm0507ba14,纯度:≥98%)、西红花苷ⅰ(批号:po4m8f30582,纯度:≥98%)、西红花苷ⅱ(批号:p23m8f36647,纯度:≥98%)上海源叶生物科技有限公司;安宫牛黄丸(北京同仁堂股份有限公司,批号:18017013;北京同仁堂科技有限公司制药厂,批号:18013301;)。天然林麝麝香样品(北京同仁堂股份有限公司提供),人工麝香(北京联鑫药业有限公司),天然牛黄样品(北京同仁堂股份有限公司提供),人工牛黄(北京同仁堂股份有限公司提供),体外培育牛黄(武汉健民大鹏药业有限公司)。
2方法与结果
2.1色谱条件
色谱柱为waterscortecsshieldrp18column(2.1mm×100mm,1.6μm),柱温38℃,流动相a乙腈-b0.3%磷酸水,梯度洗脱(0~5min,8%a;5~6min,8%~13%a;6~24min,13%~15%a;30~35min,20%~23%a;40~44min,35%~40%a;44~47min,40%~48%a;47~50min,48%~55%a;50~57min,55%~95%a;57~61min,95%~95%a;61~62min,95%~8%a),流速0.2ml/min,全波长检测。分别在检测波长为240nm(1、2、17、32、34号峰)、262nm(6号天然麝香特征峰)、274nm(8~16号色谱峰、18、20、22、23、25、28、29号色谱峰)、330nm(3、4、5、7号色谱峰)、360nm(33号天然牛黄色谱峰)、440nm(19、21、24、26、27、30、31、35号色谱峰)下分析,相对保留时间以盐酸小檗碱色谱峰为参比。
2.2供试品溶液制备
取组合物丸剂(安宫牛黄丸)适量,剪碎,精密称定,加入等量硅藻土研细,精密称定约1.5g,置于具塞三角瓶中,精密加入50ml70%乙醇,称定重量,超声(功率250w,50khz)45min,放冷,用提取溶剂补重,用0.22μm微孔滤膜过滤,取续滤液,即得供试品溶液。
取麝香样品约45mg,精密称定,置于10ml容量瓶中,加入70%乙醇,超声处理(功率250w,50khz)20min,放冷,定容至刻度,用0.22μm微孔滤膜过滤,取续滤液,即得麝香样品。
取牛黄样品约50mg,精密称定,置于10ml容量瓶中,加入70%乙醇,超声处理(功率250w,50khz)20min,放冷,定容至刻度,用0.22μm微孔滤膜过滤,取续滤液,即得牛黄样品。
2.3方法验证
精密度考察:取同一安宫牛黄丸供试品溶液,按已建立的分析方法,连续进样6针测定,记录安宫牛黄丸特征图谱中各特征图谱峰面积和保留时间,得供试品溶液特征图谱中35个特征峰保留时间和峰面积的精密度结果,各色谱峰保留时间和峰面积rsd值均小于5%,表明仪器精密度符合要求,结果见表1、表2,图1~图6。另,图7~图12显示了本实施例中典型的各检测波长下安宫牛黄丸的特征图谱。
表1特征图谱色谱峰保留时间精密度结果
表2特征图谱色谱峰峰面积值精密度结果
稳定性考察:取同一安宫牛黄丸供试品溶液,按已建立的分析方法,分别于超声处理后0、3、6、9、12、24h进样,记录特征图谱中35个特征图谱峰面积和保留时间,得供试品溶液特征图谱中35个特征峰保留时间和峰面积的稳定性结果,各色谱峰保留时间和峰面积rsd值均小于5%,表明方法稳定性符合要求,结果见表3、表4,图13~图18。表3特征图谱色谱峰保留时间稳定性结果
表4特征图谱色谱峰峰面积值稳定性结果
重复性考察:取anw样品6份,分别按样品处理方法制备样品溶液,并进样测定,记录anw特征图谱中35个特征图谱峰面积和保留时间,以盐酸小檗碱色谱峰保留时间和峰面积为参比,计算35个特征峰相对峰面积和相对保留时间,得anw供试品溶液特征图谱中35个特征峰相对峰面积和相对保留时间的重复性结果,rsd值均小于5%,表明重复性良好,结果见表5、表6,图19~图24。
表5特征图谱色谱峰相对峰面积重复性结果
表6特征图谱色谱峰相对峰面积重复性结果
方法适用性考察:选用固定色谱柱型号waterscortecsshieldrp18column(2.1mm×100mm,1.6μm),对三个不同批号色谱柱进行考察,记录anw特征图谱中35个特征图谱峰面积和保留时间。结果固定同一型号色谱柱,三个不同批号色谱柱的色谱图中35个特征图谱峰面积和保留时间rsd均小于3%。
线性关系考察:将浓度为:栀子苷136.00μg·ml-1、京尼平龙胆双糖苷30.18μg·ml-1、盐酸黄连碱27.69μg·ml-1、表小檗碱17.82μg·ml-1、盐酸小檗碱145.61μg·ml-1、盐酸巴马汀32.89μg·ml-1、汉黄芩苷30.48μg·ml-1、黄芩苷64.24μg·ml-1、黄芩素24.04μg·ml-1、汉黄芩素0.85μg·ml-1、姜黄素2.55μg·ml-1、西红花苷ⅰ12.85μg·ml-1的12个对照品混标溶液分别进样0.1、0.2、0.4、0.8、1.2、1.5、1.8μl。以进样量x(μg)为横坐标,对照品峰面积y为纵坐标,绘制标准曲线。建立色谱峰面积对进样量的回归方程见表7,结果表明,各成分在检测的浓度范围内线性关系良好。
表712个指标成分的回归方程、相关系数和线性范围
加样回收率考察:取已知指标性成分含量的本品硅藻土处理粉末6份,每份750mg,按下表中对照品加入量加入相应对照品,加入70%乙醇50ml,称定重量,超声(250w,50khz)45min,放冷,用70%乙醇溶液补足重量,用0.22μm微孔滤膜过滤,取续滤液,进样1μl测定。根据测得的各成分的量,计算各成分的加样回收率和rsd,结果见表8。12个指标性成分回收率范围92~104%,表明测定方法准确。
表8安宫牛黄丸(含硅藻土)中12个指标性成分加样回收率测定结果
实施例2、不同样品检测
分别取安宫牛黄丸处方十一味药材,及辅料蜂蜜,按照处方比例,采用已建立的样品处理方法和特征图谱分析方法进样测定,对anw特征图谱中14个特征峰进行化学成分指认,并分析35个特征峰色谱峰来源。其中包括黄连药材特征峰10个,黄芩药材特征峰9个,栀子药材特征峰9个,郁金药材特征峰4个,牛黄特征峰2个,天然麝香特征峰1个。结果见表9,图25~图28。
表9安宫牛黄丸特征图谱中35个特征峰信息
本发明的方法,可用于含天然牛黄、牛黄、天然麝香、麝香、黄芩、黄连、栀子、郁金的中药或中药组合物制剂的质量检测,通过特征峰的检验可用于天然麝香、天然牛黄及其中药组合物制剂的真伪鉴别。
- 还没有人留言评论。精彩留言会获得点赞!