一种同时测定血浆中氨基糖苷类抗生素药物浓度的HPLC-MS/MS方法与流程
本发明涉及临床血药浓度监测控制技术领域,具体地说,是一种同时测定血浆中氨基糖苷类抗生素药物浓度的hplc-ms/ms方法。
背景技术:
氨基糖苷类抗生素(aminoglyeosides),如阿米卡星、异帕米星、庆大霉素是一种由氨基糖与氨基环醇通过氧桥连接而成的苷类抗生素药物。氨基糖苷类抗生素抗菌谱广,杀菌活性强,抗生素后效应明显,因此在治疗严重的院内感染中具有相当重要的价值。然而这些抗生素主要从肾脏排泄,并有耳肾毒性,药物疗效、毒性均与血药浓度浓度相关、氨基糖苷类抗生素的优势与毒性作用都极为突出,为了保证达到有效的治疗浓度和避免不良反应,临床医生需要常监测血药浓度,结合病人病情等进行个体化给药剂量的调整,提高该类药物疗效,减少毒副反应的发生。因而,准确、灵敏的定量分析方法测定该些抗生素在血清(血浆)中的药物浓度很有必要。
现有技术中氨基糖苷类抗生素的定量方法主要有微生物法、免疫分析法、高效液相色谱法。虽然测定的方法众多,但存在测定的成本较高或者效果不理想,不能满足临床上的要求的缺陷。此外,这些方法检测标准不统一。因此,迫切需要建立一种能够同时测定上述药物的方法,这样就不必针对不同药物的检测需求来开发不同的方法,可以大幅降低后续开发成本,也有利于检测标准的统一,从而为不同检测点或实验室采集的数据能够相互参考或使用提供可能性。
中国专利申请:cn106053638b公开了一种动物源食品中氨基糖苷类抗生素残留量的检测方法,具体涉及一种基于hilic色谱柱串联三重四级杆质谱,检测动物源食品中氨基糖苷类抗生素残留量的检测方法。其特征在于:样品经过提取、固相萃取、经zic-hilic色谱柱分离后,由三重四级杆质谱测定。但该专利存与本专利检测的方法、涉及的质谱参数等条件不同,且该专利的检测下限较大。
针对上述缺陷发明人设计了本专利一种同时测定血浆中氨基糖苷类抗生素药物浓度的方法:以达比加群为内标,用含甲酸的甲醇溶液对血浆中的蛋白进行沉淀,再使用高效液相色谱分离上清液中的药物成分,然后使用高分辨质谱多反应监测模式进行药物靶向性检测,并进行定量,实现同时对血浆中三种氨基糖苷类抗生素浓度进行分析测定。本发明具有快捷性、极高的靶向性、通量高、灵敏度高、特异性强、精密度和准确度良好、稳定性好、提取回收率高、无明显基质效应和稀释效应等优点。
技术实现要素:
本发明的目的是针对现有技术的不足,提供一种同时检测血浆中三种氨基糖苷类抗生素浓度的lc-ms/ms检测方法,本方法检测时间短、灵敏度高、定量准确。
为实现上述目的,本发明采取的技术方案是:
一种同时检测血浆中三种氨基糖苷类抗生素浓度的lc-ms/ms检测方法,所述三种氨基糖苷类抗生素包括阿米卡星、异帕米星和庆大霉素,包括以下步骤:
(1)储备液的制备:用体积分数为40%-60%甲醇水溶液作为溶剂,分别配制得到一系列不同浓度的氨基糖苷类抗生素药物储备液和内标达比加群储备液;
(2)标准曲线血浆样本的制备:取空白血浆,将步骤(1)制备的不同浓度的氨基糖苷类抗生素药物储备液和内标达比加群储备液分别加入到空白血浆中,得到标准曲线血浆样本;
(3)标准曲线的设计:取步骤(2)得到的标准曲线血浆样本于试管中,涡旋振荡后加入a溶液沉淀、振荡离心,所述a溶液是按照甲酸体积:甲醇体积=0.01%-0.2%配制,然后取上清液在高效液相色谱质谱联用仪上进样分析,记录标准曲线血浆样本的色谱图,计算药物峰面积和内标峰面积的比值,以药物浓度为横坐标,以药物峰面积和内标峰面积比值为纵坐标,用加权最小二乘法进行回归运算,求得回归方程即为标准曲线;
(4)质控血浆样本的制备:取待测血浆样本,将步骤(1)得到的不同浓度的氨基糖苷类抗生素药物储备液和内标达比加群储备液分别加入到待测血浆样本中,得到一系列不同浓度的质控血浆样本;
(5)待测血浆样本中药物浓度的测定:取步骤(4)得到的不同浓度的质控血浆样本于试管中,涡旋振荡后加入a溶液沉淀、振荡离心,所述a溶液是按照甲酸体积:甲醇体积=0.01%-0.2%配制,然后取上清液在高效液相色谱质谱联用仪上进样分析,记录质控血浆样本的色谱图,计算药物峰面积和内标峰面积的比值;
(6)质控血浆样本中氨基糖苷类抗生素药物浓度的计算:根据步骤(3)得到的标准曲线来计算得到质控血浆样本中氨基糖苷类抗生素药物浓度;
所述氨基糖苷类抗生素药物为阿卡米星、异帕米星、庆大霉素;标准曲线血浆样本中各药物浓度范围如下:异帕米星0.66-20.52μg/ml;阿米卡星0.65-20.02μg/ml;庆大霉素0.75-23.00μg/ml;所述标准曲线血浆样本中内标达比加群浓度为1μg/ml。
其中,液相色谱条件:watersxbridgehilic柱(4.6×50mm,3.5μm);流动相为流动相a和流动相b,流动相梯度洗脱条件为:0-1.00min,流动相a的体积分数为90%;1.00-1.10min,流动相a的体积分数为90%-10%;1.10-4.00min,流动相a的体积分数为10%;4.00-4.10min,流动相a的体积分数为10%-90%;4.10-5.00min,流动相a的体积分数为90%。流速为0.3ml/min,进样量10ul。其中,流动相a为体积分数为0.1%的甲酸水溶液,流动相b为100%乙腈。
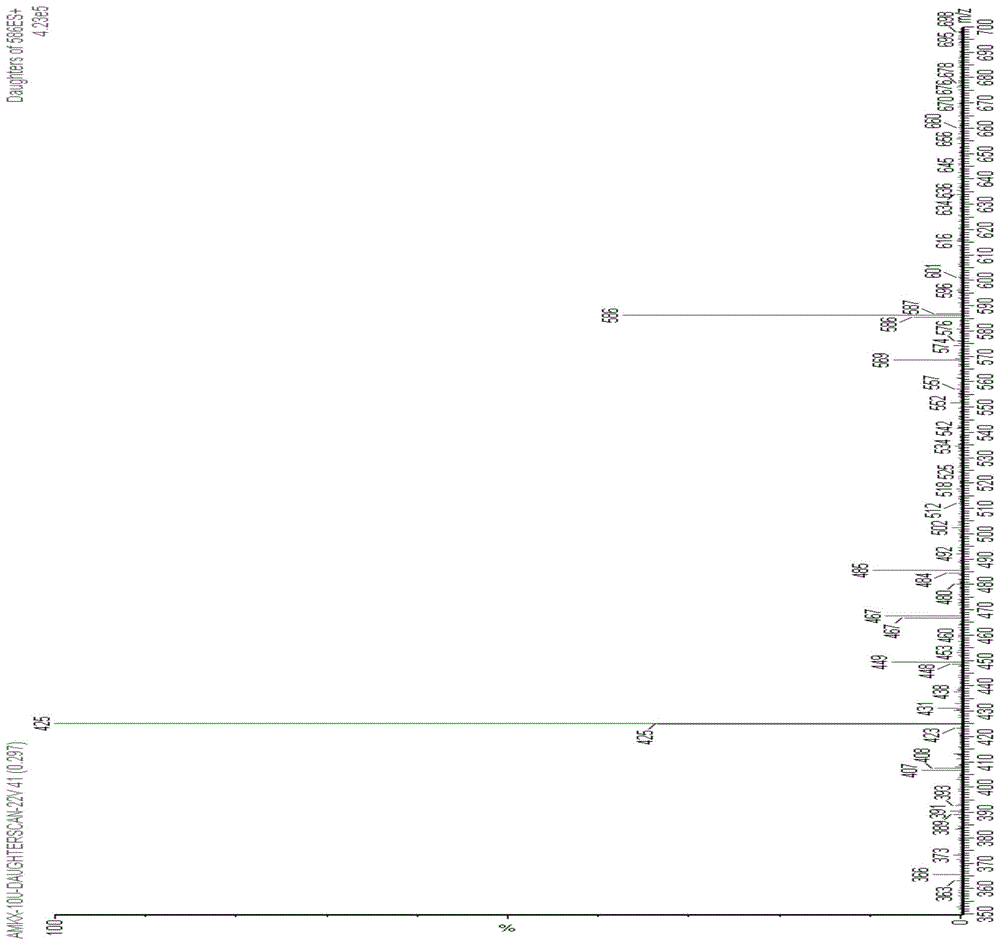
质谱条件:采用电喷雾离子源(esi),正离子检测,选择多反应监测(mrm)工作方式进行一/二级质谱分析。质谱检测工作参数如下:阿米卡星的母离子/特征子离子的检测离子对为586/425、异帕米星的母离子/特征子离子的检测离子对为570/411、庆大霉素的母离子/特征子离子的检测离子对为478/322、内标达比加群的母离子/特征子离子的检测离子对为472/289;阿米卡星的锥孔电压为22v,碰撞能量为21ev、异帕米星的锥孔电压为30v,碰撞能量为22ev、庆大霉素的锥孔电压为30v,碰撞能量为21ev、内标达比加群的锥孔电压为30v,碰撞能量为25ev;雾化气为氮气,雾化器流速为70l/hr;干燥气为氮气,干燥气温度为350℃,干燥气流速为700l/hr。
优选地,振荡离心的条件为:4℃、13000rad/min。
优选地,步骤(1)中甲醇水溶液的体积分数为50%。
优选地,所述a溶液是按照甲酸体积:甲醇体积=0.1%的方法配制。
优选地,所述待测血浆是人待测血浆。
优选地,所述标准曲线血浆样本按以下方法制得:
取90μl空白血浆,加入10μl用体积分数为50%的甲醇水稀释后的药物储备液和内标储备液,制得药物某一浓度的标准曲线血浆样本,并依此法分别制得药物的不同标准浓度的标准曲线血浆样本。其中,所述的药物储备液是以甲醇作为溶剂配制的药物浓度为1mg/ml的药物甲醇溶液;所述内标储备液是以甲醇作为溶剂配制的达比加群浓度为1mg/ml的内标甲醇溶液。
优选地,标准曲线血浆样本中最终内标浓度为1.0μg/ml,最终阿米卡星浓度分别为:20.02μg/ml、12.01μg/ml、7.21μg/ml、4.32μg/ml、2.59μg/ml、1.30μg/ml、0.65μg/ml;
异帕米星浓度分别为:20.52μg/ml、12.31μg/ml、7.39μg/ml、4.43μg/ml、2.66μg/ml、1.33μg/ml、0.66μg/ml;
庆大霉素浓度分别为:23.00μg/ml、13.80μg/ml、8.28μg/ml、4.93μg/ml、2.98μg/ml、1.49μg/ml、0.75μg/ml。
优选地,用0.1%甲酸甲醇溶液沉淀标准曲线血浆样本中蛋白,包括:将标准曲线血浆样本涡旋20s,然后加入300μl体积分数为0.1%的甲酸甲醇溶液,涡旋1min后于4℃下13000rad/min离心5分钟,取上清液100μl置进样管中,待进样分析。
优选地,用0.1%甲酸甲醇溶液沉淀待测血浆样本中蛋白,包括:将待测血浆样本涡旋20s,然后加入300μl体积分数为0.1%的甲酸甲醇溶液,涡旋1min后在4℃下13000rad/min离心5分钟,取上清液100μl置进样管中,待进样分析。
优选地,标准曲线血浆样本的进样体积为10μl。
优选地,待测血浆样本的进样体积为10μl。
本发明建立了一种基于高效液相色谱联合质谱的检测方法,先对上述血浆样品进行液相分离,随后采用质谱中的多反应监测(mrm)模式,即通过质谱中的四级杆对母离子进行选择性筛选,筛选后的离子进入碰撞池发生碰撞碎裂,产生的碎片离子进行高分辨率扫描,然后选择特征性二级碎片离子进行定量,能够有效地增强选择性,提高准确度、灵敏度。为临床上氨基糖苷类个体化治疗提供了可靠的实验数据,反馈异常结果,协助临床调整用药,在保证药物疗效的同时,可规避不良反应的发生,能指导氨基糖苷类抗生素药物的合理临床应用。
本发明优点在于:
与现有技术相比,本发明采用高效液相色谱联合质谱法同时对人血浆内的三种氨基糖苷类抗生素药物浓度进行检测,具有快捷性、极高的靶向性、通量高、灵敏度高、特异性强、精密度和准确度良好、稳定性好、提取回收率高、无明显基质效应和稀释效应的优点。
本发明方法可用于临床上常用的氨基糖苷类抗生素的血药浓度监测,指导氨基糖苷类抗生素的合理临床应用。
附图说明
附图1是阿米卡星药物的二级离子图。
附图2是异帕米星药物的二级离子图。
附图3是庆大霉素药物的二级离子图。
附图4是达比加群药物的二级离子图。
附图5是四种药物的典型图谱,其中从上到下分别为:异帕米星、庆大霉素、阿米卡星和达比加群。
具体实施方式
下面结合具体实施方式,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
实施例1
本实验用三种氨基糖苷类抗生素药物(阿卡米星、异帕米星、庆大霉素)标准品及空白血浆来制备标准曲线,并根据标准曲线来计算得到患者血浆内的药物浓度。
一、实验仪器与试剂如下:
仪器:waters2790型液质联用仪,包括waters2790高效液相,micromassquattro三重四级杆质谱仪,masslynx4.0工作站、色谱柱:watersxbridgehilic柱(4.6×50mm,3.5μm)、湘仪l550离心机、振荡器;
试剂:甲酸甲醇溶液、甲醇水溶液、阿米卡星标准品、异帕米星标准品、庆大霉素标准品、内标达比加群标准品。
二、实验方法
(一)标准曲线血浆样本和质控血浆样本的制备
1.1储备液的制备
用50%甲醇水溶液作为溶剂,分别配制三种氨基糖苷类抗生素(阿米卡星、异帕米星、庆大霉素)和内标达比加群的储备液,三种氨基糖苷类抗生素和内标达比加群的浓度均为1.0mg/ml;所有储备液均存放于-20℃冰箱。
1.2标准曲线血浆样品制备
取90μl空白血浆,加入一定体积的前述的1.1项所制得的一种药物贮备液和内标贮备液(或者加入一定体积的用体积分数为50%的甲醇水稀释后的上述1.1项所制得的一种药物贮备液和内标贮备液),制得该药物某一标准浓度的标准曲线血浆样本,并依此法分别制得各种药物的不同标准浓度的标准曲线血浆样本。标准曲线血浆样本中,最终血浆药物浓度如下表1所示,最终内标浓度为1.0μg/ml。
表1.各药物的标准曲线血浆样本的最终浓度
1.3质控血浆样品制备
取90μl空白血浆,加入一定体积的前述的1.1项所制得的一种药物贮备液和内标贮备液(或者加入一定体积的用体积分数为50%的甲醇水稀释后的上述1.1项所制得的一种药物贮备液和内标贮备液),制得该药物某一质控浓度的质控血浆样本,并依此法分别制得各个药物的不同质控浓度的质控血浆样本。质控血浆样本中,最终血浆药物浓度如下表2所示,最终内标浓度为1.0μg/ml。
表2.各药物的质控血浆样本的最终浓度
(二)实验操作
2.1血浆样品预处理
取100ul血浆样品置于1.5mlep管中,涡旋震荡10s后,加入300ul体积分数为0.1%甲酸的甲醇溶液,涡旋振荡3min,于13000r/min离心10min;分取上清液100ul,自动进样10ul进行lc-ms/ms分析,峰面积内标法定量检测。
2.2色谱条件
watersxbridgehilic柱(4.6×50mm,3.5μm);流动相为流动相a和流动相b,流动相梯度洗脱条件为:0-1.00min,流动相a的体积分数为90%;1.00-1.10min,流动相a的体积分数为90%-10%;1.10-4.00min,流动相a的体积分数为10%;4.00-4.10min,流动相a的体积分数为10%-90%;4.10-5.00min,流动相a的体积分数为90%。流速为0.3ml/min,进样量10ul。其中,流动相a为体积分数为0.1%的甲酸水溶液,流动相b为100%乙腈。
2.3质谱条件
采用电喷雾离子源(esi),正离子检测,选择多反应监测(mrm)工作方式进行一/二级质谱分析。质谱检测工作参数如下:阿米卡星的母离子/特征子离子的检测离子对为586/425、异帕米星的母离子/特征子离子的检测离子对为570/411、庆大霉素的母离子/特征子离子的检测离子对为478/322、内标达比加群的母离子/特征子离子的检测离子对为472/289;阿米卡星的锥孔电压为22v,碰撞能量为21ev、异帕米星的锥孔电压为30v,碰撞能量为22ev、庆大霉素的锥孔电压为30v,碰撞能量为21ev、内标达比加群的锥孔电压为30v,碰撞能量为25ev;雾化气为氮气,雾化器流速为70l/hr;干燥气为氮气,干燥气温度为350℃,干燥气流速为700l/hr。
(三)方法学评价
方法学验证主要包括线性、灵敏度、精密度、准确度、回收率、基质效应和稳定性。
3.1标准曲线和定量下限
对于按照“表1”中所列的某一药物的各标准浓度配制的各个标准曲线血浆样本中的每个样本进行以下操作:按照“2.1血浆样品预处理”项操作后,取10μl置于进样管中,依照“2.2”和“2.3”的条件,进行hplc-ms/ms(使用mrm模式)测定,记录色谱图;并计算药物峰面积和内标峰面积的比值;全部样本完成上述操作后,以药物浓度为横坐标,以药物峰面积和内标峰面积的比值为纵坐标,用加权(1/x2)最小二乘法进行回归运算,求得该药物的直线回归方程,即为该药物的标准曲线。依照同样的步骤,制备各药物的标准曲线。各药物的直线回归方程以及定量下限的结果如表3所示。
表3.各药物线性回归方程、相关系数、线性范围及定量下限
3.2精密度和准确度
对于按照“表2”中所列的各质控浓度配制的各药物的质控血浆样本(每个药物每个浓度各制备6份,每天制备一批样品,连续3天,一共制备3批作为考察精密度和准确度的样品)中的每个样本进行以下操作:按照“2.1血浆样品预处理”项操作后,取10μl置于进样管中,依照“2.2”和“2.3”的条件,进行hplc-ms/ms(使用mrm模式)测定,记录色谱图;并计算药物峰面积和内标峰面积的比值;代入当天所得的标准曲线求测试浓度,最后计算批内和批间精密度(绝对值小于15%为合格)和准确度(85%-115%为合格)。具体结果见表4。
表4.各药物准确度和精密度测试结果
由表4可见,血浆中各药物的各测试浓度的准确度均在85%-115%的区间内,血浆中各药物的各测试浓度的批内和批间精密度绝对值均小于15%。由此证明本发明方法对血浆中上述3个药物的检测具有良好的精密度和准确度。
3.3基质效应和绝对回收率
取6份不同来源的人血浆,按血浆前处理方法处理得空白基质,以该空白基质配制低、中、高浓度上述3个药物的质控溶液各3份进行lc-ms/ms分析。峰面积与相应浓度的药物标准溶液峰面积比值即为基质效应结果。取低、中、高血浆样品(n=5),按“2.1血浆样品预处理”项步骤操作,获得血浆中药物峰面积,与相同浓度对照品的峰面积比较,计算血浆中药物的绝对回收率。基质效应及绝对回收率结果见表5。
表5.人血浆内基质效应和提取回收率试验(n=6)
结果血浆基质对上述三个药物及内标影响程度基本一致。
3.4稀释效应(十倍稀释)
对于按照“表1”中所列的各药物的浓度7配制的各个药物血浆样本(每个药物均制备3份)分别进行以下操作:用空白血浆稀释十倍后,按照“2.1血浆样品预处理”项操作后,取10μl置于进样管中,依照“2.2”和“2.3”的条件,进行hplc-ms/ms(使用mrm模式)测定,记录色谱图;并计算药物峰面积和内标峰面积的比值;代入当天所得的标准曲线求测试浓度,将浓度结果乘以10,计算出未稀释时的血浆药物浓度。
将计算到的各药物浓度与所对应的浓度7进行比较,结果显示:10倍稀释后,各药物的精密度均在15%以内,准确度均在85%-115%之间,表明在该稀释条件下,未出现明显的稀释效应,样品的精密度和准确度不受影响。
3.5稳定性
观察血浆样品不同条件下测得的药物与内标峰面积比与血浆制备即刻(0小时)的初始峰面积比值,re%值均小于15%。血浆样品处理后进样器放置24小时、28天稳定性及冻融三次后提取均稳定,结果见表6。
表6.稳定性测试
由表6可见:血浆中各药物的各浓度在上述考察条件下检测得出的re值均小于10%,提示在上述考察条件下本发明方法对血浆中上述3个药物的检测具有较强稳定性。
本发明以达比加群为内标,先用含甲酸体积分数为0.1%的甲醇溶液对血浆中的蛋白进行沉淀,再使用高效液相色谱法分离上清液中的药物成分,然后使用高分辨质谱多反应监测模式进行药物靶向性检测,并进行定量,实现同时对血浆中三种氨基糖苷类抗生素浓度进行分析测定。具有快捷性、极高的靶向性、通量高、灵敏度高、特异性强、精密度和准确度良好、稳定性好、提取回收率高、无明显基质效应和稀释效应的优点,可很好的指导氨基糖苷类抗生素的临床合理应用。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明原理的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!