通过检测当归药材中具有钙拮抗功效的有效成分含量评价质量等级的方法与流程
本发明属于中药材品质评价的技术领域,涉及一种快速评价当归质量等级的技术方法。
背景技术:
当归为伞形科植物当归angelicasinensis(oliv.)diels的干燥根,具有补血活血,调经止痛,润肠通便之功效。当归原产于中国西部海拔较高的阴湿地区,在甘肃岷县等地及西藏地区有野生当归分布。现在市场上流通的当归均为栽培品种,主要来自甘肃、云南、湖北、四川等省区。由于受气温、土壤和种植方式等影响,各地当归性状与质量有所不同,因此对于当归的品质评价需要更便捷可靠的方法。
目前对于当归药材的指纹图谱和化学成分做了大量研究[杨英来等.中国实验方剂学杂志,2014,20(13):55-60;李国强等.中草药,2017,48(19):4017-4023],发现当归主要含挥发油、酚酸类和多糖类化合物,其中当归挥发油含量约为0.42%,油中主要含有藁本内酯约为45%,其次为正丁烯呋内酯约为11.3%[牛莉等.中西医结合心血管病杂志,2018,6(21):90-92]。当归的挥发性成分在解痉、镇痛、抗炎、抑制血小板聚集以及清除自由基和抗氧化方面具有显著的药理活性。藁本内酯是当归中可以改善血液流变性及具有强抗氧化活性的重要活性成分之一,通常被作为当归药材及其相关制剂的质量控制的指标成分[陈方等.中国药师,2018,21(10):1861-1864]。此外,当归富含具有生理活性的有机酸类化合物,虽然其结构简单,但具有极其显著的生理活性,因此,阿魏酸被广泛地认为是当归及其相关产品质量控制的指标成分[管西芹等.天然产物研究与开发,2018,30:2033-2038]。此外,当归多糖也具有丰富的生物活性,能通过激活补体对机体免疫系统、造血系统发挥作用,还具有抗肿瘤、抗放射性损伤和促进胃溃疡愈合等活性[任峰等.中国老年学杂志,2018,10(38):4749-4751]。
目前国内外沿用的中药质量评价主要方法是采用液相色谱法为主的指标成分含量测定或化学指纹图谱分析。如赵健雄等研究了当归药材的hplc指纹图谱,找出了22个共有峰,11个样品与当归对照药材之间的相似度均在90%以上,平均相似度为96.77%[赵健雄等.分析测试技术与仪器,2005,11(4):303-306]。于淼等对当归水溶性成分hplc指纹图谱进行了研究,实现了当归中多个化学成分色谱峰的分离,获取了反映药材整体的化学数据的hplc指纹图谱[于淼等.中国冶金工业医学杂志.2008,25(1):20-22]。吴燕燕等采用高效液相色谱法比较了当归饮片和全当归的指纹图谱,证明了全当归和当归饮片在化学成分上存在较大差异[吴燕燕等.j.chin.pharm.sci.2014,23(6),393-402]。王亚丽等采用高效液相色谱法对甘肃36个产地的当归药材进行了分类比较,建立了各样本的hplc共有模式图[王亚丽等.中国中药杂志,2009,34(11):1390-1394]。此外,中国专利也分别公开了采用高效液相方法检测当归中藁本内酯和阿魏酸的指纹图谱等技术[申请号:cn201810477828;cn201410375336;cn201510916633]。
2015版药典当归药材的全检项目包含了横断面、粉末的显微鉴别以及薄层检测,还对灰分、浸出物、挥发油、阿魏酸的含量进行了限定。这些指标虽然对当归的质量有一定程度的控制,但还不能够充分反映当归特定的质量属性。目前对当归质量的评价和药用功效的分析还没有有效的结合,缺乏快速简便易行的检测手段。
技术实现要素:
本发明目的是克服现有技术的不足,提供一种通过检测当归药材中具有钙拮抗功效的有效成分含量评价质量等级的方法。
本发明的技术方案概述如下:
通过检测当归药材中具有钙拮抗功效的有效成分含量评价质量等级的方法,包括如下步骤:
(1)通过超高效液相色谱法检测当归药材中z-藁本内酯和欧当归内酯a的含量,1g当归药材中z-藁本内酯的mg数用x1表示,1g当归药材中欧当归内酯a的mg数用x2表示;(2)胞内钙拮抗功效与z-藁本内酯和欧当归内酯a的关系满足下列函数:
y=(31.2579x1+381.352x2-248.979x1x2+18.4822)×100%
其中:y为当归药材胞内钙拮抗功效的评估值的简称,y的范围区间:0~100%,根据y值,将当归药材分为1级-7级七个质量等级,
y值为100~85.7%是1级;
y值为85.6~71.4%是2级;
y值为71.3~57.1%是3级;
y值为57.0~42.8%是4级;
y值为42.7~28.6%是5级;
y值为28.5~14.3%是6级;
y值为14.2~0%是7级。
本发明的优点:
本发明的方法通过“药效成分含量-钙离子拮抗活性”的关系拟合,实现了对当归药材钙拮抗功效的快速评价。该方法简便快捷,可以通过简单的uplc方法就可以实现对当归特定功效的评价。
附图说明
图1是基于ca2+拮抗活性的当归主要药效成分的uplc-q/tof谱效筛选图。
图2是当归药效成分ca2+拮抗作用的药效评价柱图。
图3是当归药效成分血管扩张作用的药效评价柱图。
图4是不同批次当归药材中主要药效成分含量测定的标准曲线与含量分布图。
图5是不同批次当归药材ca2+拮抗活性的检测图。
图6是当归关键药效成分的含量与ca2+拮抗活性的量效拟合示意图。
具体实施方式
下面结合具体实施例对本发明作进一步的说明。
实施例1
uplc-q/tof用于当归活性成分的鉴定,在用本发明的方法进行实际操作时,仅采用uplc就可以对当归活性成分进行含量测定。
1)当归药材甲醇提取物的制备:
取干燥的当归药材样本,粉碎并经100目六号筛过滤,得当归药材粉末,取其中0.5g粉末,加入50ml体积浓度70%甲醇水溶液,在功率600w,室温条件下超声提取30分钟,4000转/分钟,离心10分钟,除去残渣,取上清,经0.22μm微孔滤膜过滤,滤液置4℃保存;
2)超高效液相色谱条件:
色谱柱acquity
表1.uplc色谱洗脱条件
3)质谱条件:
离子源为软电离模式esi电喷雾离子源,数据采集工作站为watersmasslynx4.1工作站。
在正离子模式下,毛细管电压3.0kv;离子源温度110℃;锥孔电压30v;雾化气高纯氮气的流速为600l/小时,雾化器温度设定为350℃;离子质量扫描范围100~1500da,扫描频率0.1秒,扫描间隔延时0.02秒;校正液为亮氨酸脑啡肽([m+h]+=555.2931)。
在负离子模式下,毛细管电压2.5kv;离子源温度110℃;锥孔电压30v;雾化气高纯氮气的流速为600l/小时,雾化器温度设定为350℃;离子质量扫描范围100~1500da,扫描频率0.1秒,扫描间隔延时0.02秒;校正液采用亮氨酸脑啡肽([m-h]-=553.2775)。
4)进样及馏分收集
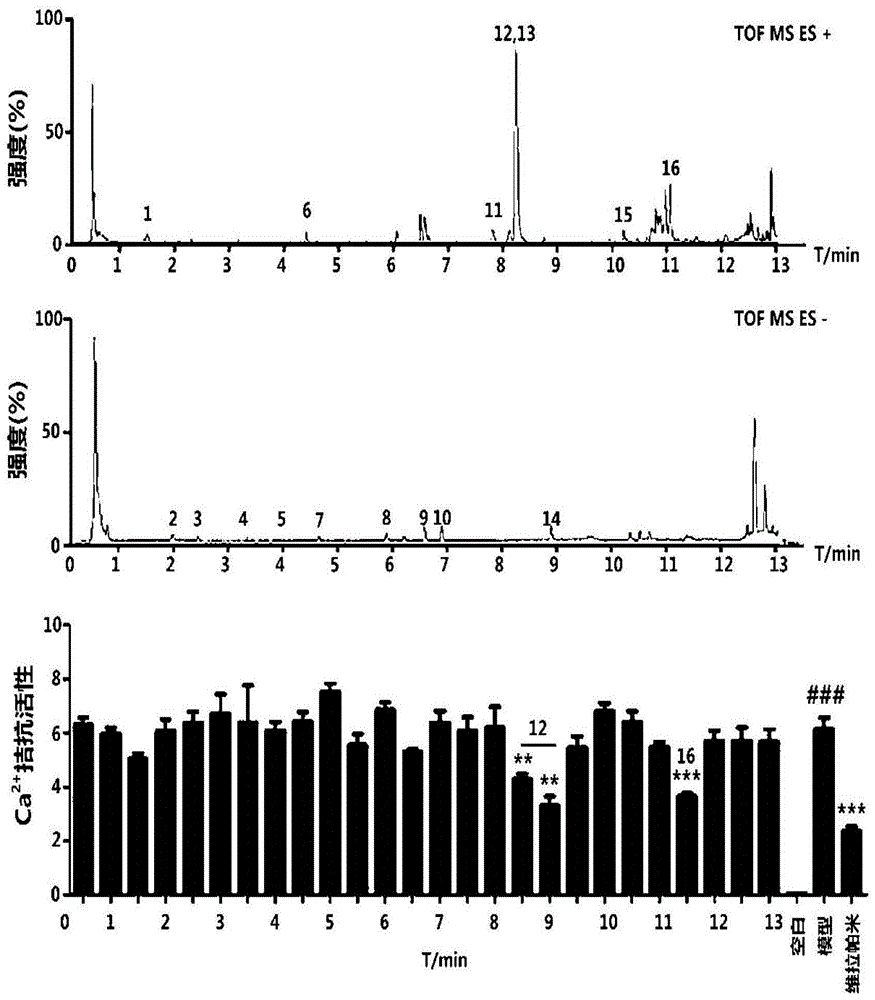
取步骤1)获得的滤液,进样10μl,经上述uplc条件分离,流出液经10:1分流,其中9/10部分每间隔30秒收集一次至96孔深孔板中,并置于真空干燥箱中减压干燥,制备uplc馏分备用。另1/10部分直接进行esi质谱检测,得到本色谱条件下的正负模式的bpi图,如图1(上、中)所示,当归药材的成分得到较好的分析,经质谱m/z解析以及与标准品的ms/ms文献比对,共标注了16个主要成分,具体见表2。
表2.当归中主要化学成分的解析
实施例2.基于ca2+拮抗活性的当归药效成分的uplc-q/tof谱效筛选
(1)细胞培养:取人肾上皮细胞293t(购于美国typeculturecollection,rockvillemd公司),加入4mldmem高糖完全培养基,置于25cm2的细胞培养瓶中,在37℃,5%co2培养箱中静置培养。待细胞生长融合度为70%时,进行细胞转染。
dmem高糖完全培养基:dmem高糖培养基(美国gibco公司)中按10%加入胎牛血清,按1%加入氨苄西林(美国gibco公司),按1%加入链霉素(美国gibco公司)。
(2)高表达质粒菌种的构建:将感受态细胞大肠杆菌dh5α(ecolidh5α)置于冰水浴中化冻,待细胞刚化冻后分别加入目的dna(ca2+荧光素酶报告pgl4.30质粒和内参荧光素酶报告基因质粒renilla)用手指拨打管底,轻轻混匀后在冰水浴中放置30分钟,取出迅速置于42℃热击90秒钟,然后迅速将其置于冰水浴中2分钟,随后加入200μl无菌的lb培养基,置于37℃摇床中,200rpm震荡复苏培养60分钟,取适量菌液涂布在含有抗性的lb平板上,倒置平板,37℃培养12h,挑取单个菌落进行扩大培养,基因组测序,分别获得高表达pgl4.30质粒和renilla内参质粒的大肠杆菌。
(3)质粒提取:将步骤(2)得到的大肠杆菌于37℃恒温摇床中扩增培养12小时,待细菌密度od为0.6时,分别提取pgl4.30质粒和renilla内参质粒,用于下列转染。
(4)细胞转染:将步骤(1)培养的293t细胞,进行细胞转染。将pgl4.30质粒(100ng/每孔)、renilla内参质粒(10ng/孔)与pei脂质体2000(美国invitrogen公司)(终浓度1mg/ml)均匀混合于dmem高糖空白培养基中,静置15分钟后,再加入上述dmem高糖完全培养基,置于37℃,5%co2培养箱中继续静置培养20小时。
dmem高糖空白培养基:dmem高糖培养基(美国gibco公司)
(5)细胞给药:实验分组为空白组(dmem高糖空白培养基),模型组(造模液),阳性组(含终浓度为10-5mol/l维拉帕米的造模液)和实验组[实施例1中所制备的当归各uplc馏分(实施例1步骤4)获得),向各uplc馏分中,加入500微升造模液],给药6小时后,收集细胞进行报告基因检测。
造模液:含终浓度为1×10-3mol/l离子霉素和1mg/ml佛波酯的dmem高糖空白培养基;
(5)报告基因检测:将给药6小时的细胞上清液弃去,并每孔分别加入双荧光素酶报告基因试剂盒(美国promega公司)中的细胞裂解液(1×)20μl(初始浓度为5×,用超纯水稀释5倍后得到1×细胞裂解液,用于裂解细胞),震荡30分钟。收集裂解液,每孔吸出15μl转移至1.5mlep管中。并按照荧光素酶报告基因试剂盒(promega,美国)的说明测定荧光强度值。依据相对荧光强度的比值(ca2+荧光值/renilla荧光值),计算ca2+抑制活性。
实验结果如图1(下)所示,模型组的细胞经药物刺激后细胞内的钙离子浓度明显增加,阳性药维拉帕米可以有效抑制其内钙的上升。同时保留时间分别在8.5~9分钟,以及11.5分钟的当归uplc分离馏分显示了明显的降钙效果。依据上述实施例1的成分鉴定解析结果,发现其对应的z-藁本内酯和欧当归内酯a为其主要的活性成分。
实施例3.当归主要活性成分z-藁本内酯和欧当归内酯a的ca2+拮抗效果验证
参照实施例1的方法,将粉碎后的当归药材过筛,取其中0.5g粉末,加入50ml体积浓度70%甲醇水溶液,在功率600w,室温条件下超声提取30分钟,4000转/分钟,离心10分钟,除去残渣,收集上清液并减压浓缩后冷冻干燥,得提取物干粉。
精密称取上述冻干粉(含z-藁本内酯:13.832mg/g和欧当归内酯a:1.476mg/g),分别以二甲基亚砜(dmso)为溶剂配制成浓度为10-1、10-2、10-3kg/l的溶液,4℃保存备用。同时分别精密称取z-藁本内酯(商品)、欧当归内酯a(商品)适量,经少量dmso溶解后配制成浓度为10-1、10-2、10-3mol/l的标准溶液,-20℃保存备用。再按照z-藁本内酯和欧当归内酯a在当归冻干粉中的含量比例,精密称取z-藁本内酯1.38mg和欧当归内酯a0.15mg,溶于1mldmso中制成10-1kg/l混标溶液,在此基础上用dmso稀释10倍、100倍得到10-2、10-3kg/l的混标溶液,于-20℃保存备用。
实验分组为空白组(dmem高糖空白培养基),模型组(造模液),阳性组(含终浓度为10-5mol/l维拉帕米的造模液)和上述所制备的当归提取物组(分别用造模液稀释上述冻干粉溶液,使其终浓度为10-4、10-5、10-6kg/l),和z-藁本内酯、欧当归内酯a标准品组,以及混标组(分别用造模液稀释上述混标溶液,使其终浓度为10-4、10-5、10-6kg/l),给药6小时后弃去细胞上清液,并每孔分别加入1×细胞裂解液20μl,震荡30分钟。收集裂解液,每孔吸出15μl转移至1.5mlep管中。并按照荧光素酶报告基因试剂盒(promega,美国)的说明测定荧光强度值。依据相对荧光强度的比值(ca2+荧光值/renilla荧光值),计算ca2+抑制活性。
实验结果如图2所示,z-藁本内酯、欧当归内酯a浓度依赖地展示了ca2+拮抗作用。并且z-藁本内酯与欧当归内酯a的混标组与当归提取物组对比没有显著的差异,再次说明可以通过采用关键活性化合物z-藁本内酯、欧当归内酯a的含量来评价当归的整体ca2+拮抗作用。
实施例4.当归药效成分z-藁本内酯和欧当归内酯a的血管扩张作用的评价
取体重200±20g健康的sd大鼠,处死后迅速打开胸腔取出胸主动脉,立即放入4℃预冷的k-h液中(nacl118mmol/l,kcl4.7mmol/l,nahco325mmol/l,kh2po41.2mmol/l,mgcl21.2mmol/l,cacl22.5mmol/l,葡萄糖2g/l,ph7.4)。小心去除动脉周围结缔组织后,用玻璃棒轻轻碾压血管,去除内皮,并剪成长约3毫米的动脉环。将处理好的动脉环挂在张力传感器上,置于37℃恒温浴槽中,持续通入95%o2和5%co2的混合气体,使动脉环处于松弛状态,固定,并调整动脉环张力至0.5g,持续15分钟,换液,基线上调0.5g,持续15分钟,再换液,再上调,使大动脉调整动脉环张力逐步上调至2.0g,稳定1.5小时后开始实验。
首先加入1mmol/l高钾溶液(称取kcl3.7g,溶解在50ml超纯水中,即得1mol/l的高钾溶液,取10μl加入到10mlk-h液中即得1mmol/l的高钾溶液)检测血管活性,若血管活性良好,再后加入10-6mol/l的去甲肾上腺素水溶液10μl,刺激血管收缩4~6分钟。待血管收缩至平台期后,加入浓度为10-4mol/l的乙酰胆碱水溶液10μl作用4分钟,检测血管内皮的完整性(舒张率小于10%即认为血管内皮去除完全)。换液,加入10ml的k-h液,平衡张力到2.0g,稳定15分钟。再加入终浓度10-6mol/l去甲肾上腺素10μl,刺激血管收缩(4~6分钟),待血管收缩到平台期后,分别向各个通道加入相应药物,空白组加入等量dmso的溶液,阳性药组加入终浓度为10-5mol/l硝苯地平,实验组分别加入10-1、10-2、10-3mol/lz-藁本内酯、欧当归内酯a标准品溶液、混标溶液以及当归提取物溶液10μl,作用8~10分钟,通过lab-chart7多通道生理记录观察给药前后张力变化,每批重复3次,计算其舒张率[舒张率=(平台期张力-舒张后张力)/(平台期张力-2.0g)]。实验结果如图3所示,z-藁本内酯、欧当归内酯a展示了浓度依赖性的血管舒张作用,而与当归提取物相比,以同样计量的z-藁本内酯和欧当归内酯a的混标其血管舒张作用没有显著差异,再次说明可以通过采用z-藁本内酯和欧当归内酯a的含量,来评价当归的血管扩张作用。
实施例5.不同批次当归药材中z-藁本内酯和欧当归内酯a的uplc含量分析
样品的制备方法和uplc检测方法参照实施例1进行。分别精密称取z-藁本内酯11.27mg和欧当归内酯a1.12mg于50ml容量瓶中,用体积浓度为70%甲醇水溶液溶解并稀释至刻度,摇匀,0.22μm微孔滤膜过滤,分别精密量取续滤液0.8、1、1.5、2、2.5、3、4、5、6μluplc进样分析,计算测定对应的峰面积,以各色谱峰峰面积积分值(y)对质量(x)进行线性回归,绘制标准曲线。实验结果如图4(上),所得到z-藁本内酯(左)和欧当归内酯a(右)的标准曲线具有良好的线性,r2均大于0.9998。
选取50批不同产地的当归药材,分别粉碎并过五号筛(80目),取其中0.5g粉末,加入50ml体积浓度为70%甲醇水溶液,室温下超声30分钟,4000转/分钟,离心10分钟,分别取上清液500μl于1.5mlep管中,真空干燥后,4℃保存备用。
以实施例1的uplc检测方法为基础,分别对上述当归提取物进行检测分析,计算其相对含量。结果如图4(下)所示,50批当归药材中z-藁本内酯的含量范围(左)为:2.248~12.303mg/g,其均值为:5.853mg/g。欧当归内酯a的含量范围(右)为:0.1105~0.7607mg/g,其均值:0.3016mg/g。
实施例6.不同当归批次样品ca2+拮抗活性的评价
随机选取上述实施例5中保存备用的当归样本14个批次。实验分组为空白组、模型组、阳性组和当归提取物组(上述样品中加入500μldmem高糖空白培养基,室温超声30分钟,溶解),每个批次分别用1×10-3mol/l离子霉素和1mg/ml佛波酯配置而成的造模液稀释5、10、20、40倍四个不同的浓度梯度,给药6小时后,收集细胞,弃去细胞上清液,并每孔分别加入1×细胞裂解液20μl,震荡30分钟。收集裂解液,每孔吸出15μl转移至1.5mlep管中。并按照荧光素酶报告基因试剂盒(promega,美国)的说明测定荧光强度值。依据相对荧光强度的比值(ca2+荧光值/renilla荧光值),计算ca2+抑制活性。
实验结果图5所示,56组不同批次和浓度的当归提取物呈现出不同的ca2+拮抗效果,提示不同的药效成分的含量与配比关系对整体活性有着不同的影响。
实施例7.当归主要药效物质含量与ca2+拮抗活性的关系拟合
为了建立z-蒿本内酯(x1)、欧当归内酯a(x2)的含量与y(当归药材胞内钙拮抗功效的评估值)之间的函数关系,通过建立二元二项式方程拟合函数关系,并使用非线性规划方法最优化z-藁本内酯与欧当归内酯a之间的最佳含量组合。
具体而言,设定函数关系方程为
y=(a,x)a(1)+a(2)x1+a(3)x2+a(4)x1x2+a(5)x12+a(6)x22
其中a(1)~a(6)为待优化的参数。
将上述实施例6的56组活性与其实施例5中对应的含量带入y函数内,其中x1∈(0.0906~2.3241‰),x2∈(0.0028~0.1521‰)。
使用逐步回归策略,筛选f1函数中的重要变量,设定entrancetolerance为0.05,即在建模过程中根据f检验的p值,入选变量的p<0.05;设定exittolerance为0.1,经过反复的引入剔除变量简单函数关系。
最终得到的活性评价函数关系如下:
y=(31.2579x1+381.352x2-248.979x1x2+18.4822)×100%
在此公式下所建立的模型显著性强,f=44.9811,p=1.77846×10-14,数据拟合的回归系数r2达到0.72,说明建立好的数学模型可用于当归药材的活性进行预测,y为当归药材整体的胞内钙拮抗功效的评估值的简称,y的范围区间:0~100%,根据y值,将当归药材分为1级-7级七个质量等级,y值越大则钙拮抗活性越强。y为100~85.7%是1级;y为85.6~71.4%是2级;y为71.3~57.1%是3级;y为57.0~42.8%是4级;y为42.7~28.6%是5级;y为28.5~14.3%是6级;y为14.2~0%是7级。
实施例8.通过检测当归药材中具有钙拮抗功效的有效成分含量评价质量等级的方法
(1)取50批甘肃产的当归药材,粉碎,过药用筛的七号筛(120目),分别准确称取上述药材粉末0.5g,加入50ml70%(7:3,v/v)甲醇水溶液,室温下超声30分钟,4000转/分钟,离心10分钟,分别取上清液500μl,用0.22μm微孔滤膜过滤,参照实施例1中的液相条件,通过uplc含量分析检测50批当归药材中的z-藁本内酯和欧当归内酯a的含量(见表3,即1g当归药材中z-藁本内酯的mg数用x1表示,1g当归药材中欧当归内酯a的mg数用x2表示)。
(2)胞内钙拮抗功效与z-藁本内酯和欧当归内酯a的关系满足下列函数(进一步将所拟合分析的z-藁本内酯和欧当归内酯a的含量导入实施例7中的函数方程式):
y=(31.2579x1+381.352x2-248.979x1x2+18.4822)×100%
分别计算50批当归药材胞内钙拮抗功效(y值),并建立z-蒿本内酯(x1)、欧当归内酯a(x2)的含量与y之间的关联关系。结果如表3所示,依据药效成分的含量不同,得出50批样品的y,并按照y值大小将不同批次的样品分为不同的质量等级。因此通过该方法实现了当归药材扩张血管的功效强弱的快速评价以及品质等级的区分。
表350批当归样品扩血管功效等级预测
- 还没有人留言评论。精彩留言会获得点赞!