一种用于痰喘药物组合物的质量检测方法与流程
本发明涉及医药发明领域,具体涉及一种用于痰喘药物组合物的质量检测方法。
背景技术:
痰喘半夏颗粒是制半夏、川贝母、肉桂、豆蔻、沉香、丁香、西洋参、白芷、细辛、天竺黄、朱砂、甘草、陈皮、川芎、麸炒枳壳、麸炒白术、麸炒青皮、泽泻、麸炒白芍、山楂、制五味子、酸枣仁、干姜、薄荷油制成,具有止咳,化痰,平喘的作用,用于新老咳嗽,痰多气喘。
痰喘半夏颗粒原有的质量标准(ws3-b-2257-96)只对水分及颗粒剂项下有关的各项规定进行检查,质量检测方法较简单,难以对产品的质量实现有效的控制,因此,为了更好的检测痰喘半夏颗粒质量情况,进一步提升和控制痰喘半夏颗粒的质量,本发明新增显微鉴别、白芍的薄层鉴别、白芷的薄层鉴别、五味子的薄层鉴别、沉香的液相鉴别、对马兜铃酸a进行了限量检查、白芍、甘草、五味子、沉香的hplc含量测定。
技术实现要素:
针对现有的技术问题,本发明的目的是提供一种用于痰喘的药物组合物的质量检测方法。
本发明所述的一种用于痰喘的药物组合物的质量检测方法,该组合物由制半夏、川贝母、肉桂、豆蔻、沉香、丁香、西洋参、白芷、细辛、天竺黄、朱砂、甘草、陈皮、川芎、麸炒枳壳、麸炒白术、麸炒青皮、泽泻、麸炒白芍、山楂、制五味子、酸枣仁、干姜、薄荷油制成,具体质量检测方法为:对产品进行显微鉴别,利用薄层色谱法对产品中的芍药苷、欧前胡素、五味子醇甲、斯皮诺素进行薄层鉴别,利用高效液相色谱法对产品中的沉香四醇进行鉴别,利用高效液相色谱法对产品中的马兜铃酸a的含量进行检查,利用高效液相色谱法测定产品中的芍药苷含量、沉香四醇含量、甘草苷和甘草酸的含量。
本发明所述对产品进行显微鉴别的方法为:取本品研细,粉末用水溶解,过滤,取滤渣透化或直接用水装片置显微镜下观察,具体特征如下:
产品中制半夏的显微特征:淀粉粒圆形或类圆形,直径可至15~25μm,脐点多居中,呈短缝状、人字状,偏光下观察,脐点处汇聚的消光呈较对称的十字形;草酸钙针晶束,多断裂,长可至200~260μm;
产品中川贝母的显微特征:淀粉粒广卵形、长椭圆形、贝壳形、肾形,直径可至38~46μm,脐点点状、短缝状、人字状、星状,多位于较小端,偏光下观察,脐点处汇聚的消光常呈不对称的十字形,或呈л形和乂形;
产品中肉桂的显微特征:石细胞散在,多破碎,完整者呈类方形、类长方形、类圆形、圆多角形、椭圆形,直径32~60μm,壁厚,有的一面稍尖突,有的一面菲薄;纤维梭形,木化,边缘微波状,多断裂,长可至120~180μm,直径至40~60μm,璧极厚,孔沟不明显或稀少而细短,胞腔线形;
产品中豆蔻的显微特征:胚乳细胞淀粉团,长类方形、类圆形、不规则形,长可至120~180μm,由众多细小淀粉粒集结而成;
产品中沉香的显微特征:具缘纹孔导管碎片可见,具缘纹孔呈蜂窝状紧密排列;
产品中朱砂的显微特征:不规则颗粒和碎片散在,大小不一,鲜红色或暗红色,部分具光泽。
本发明所述利用薄层色谱法对产品中的芍药苷进行薄层鉴别的方法为:
取本品15~25g,研细,加乙醇40~60ml,超声处理20~40分钟,滤过,滤液蒸干,残渣加水15~25ml使溶解,用水饱和正丁醇萃取2~4次,每次15~25ml,合并正丁醇液,用氨水洗涤2~4次,每次15~25ml,弃去氨水,取正丁醇液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液,另取白芍对照药材0.5~1.5g,加乙醇15~25ml,同法制成对照药材溶液,再取芍药苷对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液,照通则0502薄层色谱法试验,吸取供试品溶液、对照药材溶液各15μl,对照品溶液10μl,分别点于同一硅胶g薄层板上,以比例为40:5:7:0.2的二氯甲烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至显色清晰,在日光下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
本发明所述利用薄层色谱法对产品中的欧前胡素进行薄层鉴别的方法为:取本品15~25g,研细,加乙醇20~40ml,超声处理15~25分钟,滤过,滤液蒸干,残渣加水15~25ml使溶解,用60℃~90℃石油醚萃取2~4次,每次20~40ml,合并石油醚液,蒸干,残渣加60℃~90℃石油醚1ml使溶解,作为供试品溶液,另取白芷对照药材1g,加乙醇15~25ml,同法制成对照药材溶液;再取欧前胡素对照品,加甲醇制成每1ml含0.04mg的溶液,作为对照品溶液;照通则0502薄层色谱法试验,吸取供试品溶液8μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以比例为10:5:0.2的环己烷-乙酸乙酯-甲醇为展开剂,展开,取出,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
本发明所述利用薄层色谱法对产品中的五味子醇甲进行薄层鉴别的方法为:取本品15~25g,加水40~60ml使溶解,离心5~15分钟,取上清液,置分液漏斗中,用水饱和正丁醇振摇提取2~4次,每次20~40ml,合并正丁醇液,用正丁醇饱和的水洗涤2~4次,每次20~40ml,取正丁醇液蒸干,残渣加甲醇1ml使溶解,置中性氧化铝柱上,用甲醇20~40ml洗脱,收集洗脱液,浓缩至1ml作为供试品溶液;另取五味子对照药材1g,加甲醇10~30ml,超声处理10~30分钟,滤过,滤液浓缩至1ml,作为对照药材溶液;再取五味子醇甲对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液;照通则0502薄层色谱法试验,吸取供试品溶液5μl,对照药材溶液和对照品溶液各3μl,分别点于同一硅胶gf254薄层板上,以比例为12:4:0.4的甲苯-乙酸乙酯-冰醋酸为展开剂,展开,取出,晾干,在254nm紫外灯下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
本发明所述中性氧化铝柱具体为中性氧化铝量为5g,过100~200目的筛,内径1cm,干法装柱。
本发明所述利用薄层色谱法对产品中的斯皮诺素进行薄层鉴别的方法为:取本品5~15g,研细,加甲醇40~60ml,超声处理20~40分钟,滤过,滤液蒸干,残渣加水3~9ml使溶解,置spec18小柱6cc上,依次用水3~9ml,35~45%的甲醇溶液3~9ml洗脱,弃去洗脱液,再用80~90%甲醇溶液3~9ml洗脱,收集洗脱液,洗脱液浓缩至1ml,作为供试品溶液;另取酸枣仁对照药材1g,加甲醇15~25ml,超声处理20~40分钟,滤过,滤液蒸干,加1ml甲醇使溶解,作为对照药材溶液,再取斯皮诺素对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液,照通则0502薄层色谱法试验,吸取供试品溶液15μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以水饱和正丁醇为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
本发明所述利用薄层色谱法对产品中的沉香四醇进行薄层鉴别的方法为:取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入60~80%甲醇溶液15~25ml,称重,功率720w,频率40khz条件下超声处理20~40分钟,取出,再称重,用60~80%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;另取沉香对照药材0.5g,加60~80%甲醇溶液5~15ml,超声处理20~40分钟,滤过,取续滤液作为对照药材溶液,再取沉香四醇对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液,照通则0512高效液相色谱法试验,分别精密吸取供试品溶液、对照品溶液、对照品溶液各20μl,注入液相色谱仪,记录;供试品色谱图中,应呈现与沉香四醇对照品保留时间相一致的色谱峰,并同时呈现与对照药材中另一特征峰(其相对于沉香四醇的保留时间为2.95±1%)保留时间相一致的色谱峰。
所述高效液相色谱条件具体为:
采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长250nm,理论塔板数按芍药苷峰计算,应不低于15000;
本发明所述利用高效液相色谱法对产品中的马兜铃酸a的含量检查方法为:
马兜铃酸a照高效液相色谱法测定;
色谱条件以十八烷基硅烷键合硅胶为填充剂;以比例为38:62的乙腈~0.5%磷酸溶液为流动相;检测波长为270~350nm;理论塔板数按马兜铃酸a峰计算,应不低于30000;
对照品溶液的制备取马兜铃酸a对照品,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,摇匀,即得;
供试品溶液的制备取本品20袋,混匀,研细,取约20g,精密称定,置锥形瓶中,精密加入甲醇100~200ml,称重,摇匀,在功率720w,频率40khz条件下超声处理20~40分钟,取出,再称重,用甲醇补足减失重量,摇匀,滤过;精密吸取续滤液80~120ml,蒸干,残渣加0.5%氢氧化钠溶液20~40ml溶解,分次转移到分液漏斗中,用三氯甲烷萃取2~4次,每次20~30ml,弃去三氯甲烷液;水层用盐酸调ph至2,用三氯甲烷萃取2~5次,每次20~30ml,合并三氯甲烷液,蒸干,残渣用甲醇溶解并转移到10ml容量瓶中,加甲醇至刻度,摇匀,离心,取上清液,作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含马兜铃酸ac17h11no7,不得过1.00μg;
本发明所述重金属及有害元素检测照铅、镉、汞、砷、铜测定法(通则2321)测定,本品每袋含hg应为20.2~33.8mg。
本发明所述利用高效液相色谱法测定产品中的芍药苷含量、沉香四醇含量、甘草苷和甘草酸的含量的方法为:
含量测定白芍、甘草、沉香照通则0512高效液相色谱法测定;
色谱条件与系统适用性试验采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按规定进行梯度洗脱;芍药苷检测波长210~255nm、沉香四醇、甘草酸检测波长为210~310nm、甘草苷检测波长为215~317nm,理论塔板数按芍药苷峰计算,应不低于15000,所述梯度洗脱具体规定为:
对照品溶液的制备取芍药苷、沉香四醇、甘草苷、甘草酸对照品适量,加70%甲醇溶液制成每1ml含芍药苷200μg、沉香四醇40μg、甘草苷100μg和甘草酸80μg的混合溶液,摇匀,即得;
供试品溶液的制备取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入50~90%甲醇溶液5~30ml,称重,功率720w,频率40khz条件下超声处理30~60分钟,取出,再称重,用60~80%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含白芍以芍药苷c23h28o11计,不得少于4.3mg;含沉香以沉香四醇c17h18o6计,不得少于1.0mg;含甘草以甘草苷c21h22o9和甘草酸c42h62o16计,分别不得少于2.0mg和1.8mg。
本发明所述药物组合物的质量检测方法具体为:
(一)鉴别:
(1)取本品研细,粉末用水溶解,过滤,取滤渣透化或直接用水装片置显微镜下观察,具体特征如下:
产品中制半夏的显微特征:淀粉粒圆形或类圆形,直径可至20μm,脐点多居中,呈短缝状、人字状,偏光下观察,脐点处汇聚的消光呈较对称的十字形;草酸钙针晶束,多断裂,长可至230μm;
产品中川贝母的显微特征:淀粉粒广卵形、长椭圆形、贝壳形、肾形,直径可至43μm,脐点点状、短缝状、人字状、星状,多位于较小端,偏光下观察,脐点处汇聚的消光常呈不对称的十字形,或呈л形和乂形;
产品中肉桂的显微特征:石细胞散在,多破碎,完整者呈类方形、类长方形、类圆形、圆多角形、椭圆形,直径32~60μm,壁厚,有的一面稍尖突,有的一面菲薄;纤维梭形,木化,边缘微波状,多断裂,长可至150μm,直径至50μm,璧极厚,孔沟不明显或稀少而细短,胞腔线形;
产品中豆蔻的显微特征:胚乳细胞淀粉团,长类方形、类圆形、不规则形,长可至150μm,由众多细小淀粉粒集结而成;
产品中沉香的显微特征:具缘纹孔导管碎片可见,具缘纹孔呈蜂窝状紧密排列;
产品中朱砂的显微特征:不规则颗粒和碎片散在,大小不一,鲜红色或暗红色,部分具光泽;
(2)取本品20g,研细,加乙醇50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水20ml使溶解,用水饱和正丁醇萃取2次,每次20ml,合并正丁醇液,用氨水洗涤3次,每次20ml,弃去氨水,取正丁醇液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液;另取白芍对照药材1g,加乙醇20ml,同法制成对照药材溶液;再取芍药苷对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法试验,吸取供试品溶液、对照药材溶液各15μl,对照品溶液10μl,分别点于同一硅胶g薄层板上,以比例为40:5:7:0.2的二氯甲烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至显色清晰,在日光下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;
(3)取本品20g,研细,加乙醇30ml,超声处理20分钟,滤过,滤液蒸干,残渣加水20ml使溶解,用60℃~90℃石油醚萃取2次,每次30ml,合并石油醚液,蒸干,残渣加60℃~90℃石油醚1ml使溶解,作为供试品溶液;另取白芷对照药材1g,加乙醇20ml,同法制成对照药材溶液;再取欧前胡素对照品,加甲醇制成每1ml含0.04mg的溶液,作为对照品溶液;照薄层色谱法试验,吸取供试品溶液8μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以比例为10:5:0.2的环己烷-乙酸乙酯-甲醇为展开剂,展开,取出,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(4)取本品20g,加水50ml使溶解,离心10分钟,取上清液,置分液漏斗中,用水饱和正丁醇振摇提取2次,每次30ml,合并正丁醇液,用正丁醇饱和的水洗涤3次,每次30ml,取正丁醇液蒸干,残渣加甲醇1ml使溶解,置中性氧化铝柱(5g,100~200目,内径1cm,干法装柱)上,用甲醇30ml洗脱,收集洗脱液,浓缩至1ml作为供试品溶液;另取五味子对照药材1g,加甲醇20ml,超声处理20分钟,滤过,滤液浓缩至1ml,作为对照药材溶液;再取五味子醇甲对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液;照薄层色谱法(通则0502)试验,吸取供试品溶液5μl,对照药材溶液和对照品溶液各3μl,分别点于同一硅胶gf254薄层板上,以比例为12:4:0.4的甲苯-乙酸乙酯-冰醋酸为展开剂,展开,取出,晾干,在254nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(5)取本品10g,研细,加甲醇50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水6ml使溶解,置spec18小柱6cc上,依次用水6ml,40%的甲醇溶液6ml洗脱,弃去洗脱液,再用90%甲醇溶液6ml洗脱,收集洗脱液,洗脱液浓缩至1ml,作为供试品溶液;另取酸枣仁对照药材1g,加甲醇20ml,超声处理30分钟,滤过,滤液蒸干,加1ml甲醇使溶解,作为对照药材溶液;再取斯皮诺素对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法试验,吸取供试品溶液15μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以水饱和正丁醇为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(6)取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇溶液20ml,称重,功率720w,频率40khz条件下超声处理30分钟,取出,再称重,用70%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;另取沉香对照药材0.5g,加70%甲醇溶液10ml,超声处理30分钟,滤过,取续滤液作为对照药材溶液;再取沉香四醇对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液;照高效液相色谱法试验,分别精密吸取供试品溶液、对照品溶液、对照品溶液各20μl,注入液相色谱仪,记录;供试品色谱图中,应呈现与沉香四醇对照品保留时间相一致的色谱峰,并同时呈现与对照药材中另一特征峰保留时间相一致的色谱峰;
所述高效液相色谱条件具体为:
采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按以下的规定进行梯度洗脱;检测波长250nm,理论塔板数按芍药苷峰计算,应不低于15000;
(二)检查
马兜铃酸a照通则0512高效液相色谱法测定;
色谱条件以十八烷基硅烷键合硅胶为填充剂;以比例为38:62的乙腈~0.5%磷酸溶液为流动相;检测波长为320nm;理论塔板数按马兜铃酸a峰计算,应不低于30000;
对照品溶液的制备取马兜铃酸a对照品,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,摇匀,即得;
供试品溶液的制备取本品20袋,混匀,研细,取约20g,精密称定,置锥形瓶中,精密加入甲醇150ml,称重,摇匀,功率720w,频率40khz条件下超声处理30分钟,取出,再称重,用甲醇补足减失重量,摇匀,滤过;精密吸取续滤液100ml,蒸干,残渣加0.5%氢氧化钠溶液30ml溶解,分次转移到分液漏斗中,用三氯甲烷萃取3次,每次25ml,弃去三氯甲烷液;水层用盐酸调ph至2,用三氯甲烷萃取4次,每次25ml,合并三氯甲烷液,蒸干,残渣用甲醇溶解并转移到10ml容量瓶中,加甲醇至刻度,摇匀,离心,取上清液,作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含马兜铃酸ac17h11no7,不得过1.00μg;
重金属及有害元素照通则2321铅、镉、汞、砷、铜测定法测定,本品每袋含hg应为20.2~33.8mg。
(三)含量测定
(1)白芍、甘草、沉香照高效液相色谱法测定;
色谱条件与系统适用性试验采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;芍药苷检测波长235nm、沉香四醇、甘草酸检测波长250nm、甘草苷检测波长272nm;理论塔板数按芍药苷峰计算,应不低于15000;
对照品溶液的制备取芍药苷、沉香四醇、甘草苷、甘草酸对照品适量,加70%甲醇溶液制成每1ml含芍药苷200μg、沉香四醇40μg、甘草苷100μg和甘草酸80μg的混合溶液,摇匀,即得;
供试品溶液的制备取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇溶液20ml,称重,功率720w,频率40khz的条件超声处理30分钟,取出,再称重,用70%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含白芍以芍药苷c23h28o11计,不得少于4.3mg;含沉香以沉香四醇c17h18o6计,不得少于1.0mg;含甘草以甘草苷c21h22o9和甘草酸c42h62o16计,分别不得少于2.0mg和1.8mg。
本发明所述的通则指2015年版中国药典第四部通则。
本发明所述每袋的量为3g。
本发明用法与用量:吞服或冲服一次3g,一日2次;重者一日3次。
本发明与现有技术相比具有以下优点:
1、与现有技术相比,痰喘半夏颗粒现行的质量标准(ws3-b-2257-96)只对水分及颗粒剂项下有关的各项规定进行检测,质量检测方法较简单,难以对产品的质量实现有效的控制,而本发明新增显微鉴别、白芍的薄层鉴别、白芷的薄层鉴别、五味子的薄层鉴别、沉香的液相鉴别、对马兜铃酸a进行了限量检查、白芍、甘草、五味子、沉香的hplc含量测定,进一步提升和控制痰喘半夏颗粒的质量。
2、本发明提供了一种有较为全面的有效成分稳定性的定性、定量检测方法。
3、通过对新增白芍的薄层鉴别方法的验证,使用本发明的检测方法后供试品、对照药材、对照品、阴性样品均斑点清晰,且阴性无干扰;通过耐用性试验-不同厂家硅薄层板验证色谱效果重现性良好;通过专属性试验阴性溶液在芍药苷斑点相应位置上未出现斑点,说明专属性良好。
4、通过对新增白芷的薄层鉴别验证,使用本发明后样品在欧前胡素对照品和对照药材相应位置上均出现相对应的荧光斑点,且阴性无干扰;通过不同点样量试验,主斑点的大小适中且清晰;通过耐用性试验-不同厂家硅薄层板验证,色谱效果重现性良好;通过专属性试验阴性溶液在欧前胡素斑点相应位置上未出现斑点,在白芷对照药材色谱图的相应位置上也未出现相应斑点,说明专属性良好。
5、通过对新增五味子的薄层鉴别验证,对展开剂的筛选试验,本发明使用的展开剂中供试品、对照品、对照药材、阴性样品均斑点清晰,且阴性无干扰;通过耐用性试验-不同厂家硅薄层板验证色谱效果重现性良好;通过专属性试验,阴性溶液在五味子醇甲斑点相应位置上未出现斑点,说明专属性良好。
6、通过对新增酸枣仁的薄层鉴别验证,样品在斯皮诺素对照品和酸枣仁对照药材的相应位置上均出现相应的荧光斑点,通过耐用性试验-不同厂家硅薄层板验证,色谱效果重现性良好;通过专属性试验,阴性溶液在斯皮诺素斑点相应位置上未出现相应斑点,在酸枣仁对照药材溶液的相应位置上也未出现相应斑点,说明专属性良好。
7、通过对新增沉香的液相鉴别验证,使用本发明供试品溶液色谱图中检出了沉香四醇和沉香特征峰,且阴性未出现该二个峰,表明无干扰。
8、本发明通过马兜铃酸a的限量检查,通过精密度试验,马兜铃酸a色谱峰峰面积的平均值为4457,rsd为0.08%(n=6),表明仪器精密度良好;通过线性关系考察,回归方程为y=2452x-6.111,r=0.99999(n=6),马兜铃a在0.1239~3.717μg范围内线性关系良好;通过专属性试验,缺细辛的阴性样品在马兜铃酸a相应保留时间区域未出现色谱峰,表明阴性样品对供试品检测无干扰;通过稳定性试验,马兜铃酸a的峰面积值的平均值3866,rsd为0.53%(n=6),表明溶液在24h内稳定;通过重复性试验,6份样品中均未检测到马兜铃酸a峰;通过加样回收率试验,平均回收率85.40%(药典规定0.01%含量的回收率应在85%-110%),rsd为1.55%(n=6);通过检测限试验,马兜铃酸a的检测限=0.1031μg/μl*1/100*1/20*10μl*1000=0.5155ng;通过耐用性试验,供试品中均未检出马兜铃酸a。
9、本好明对痰喘半夏颗粒中马兜铃酸a限度的制定,最终按照更严的日服量来制定痰喘半夏颗粒中马兜铃酸a限量为不得过1.00μg/袋,服用安全,无毒副作用。
10、本品中的朱砂有毒,不宜大量服用,在药典中的最大日服量为0.5g,因此以朱砂中hg含量84.5%计算则hg的日服量不得过0.43g。本品最大日服量为9g,本品严格按照朱砂处方量投料,则本品中hg的日服量为84.5%*13.333g/1000g*9g=0.1014g/天(相当于本品中hg为33.8mg/袋),远低于药典规定的0.43g/天(相当于本品中hg为0.143g/袋)。表明本品中朱砂的处方量以及在规定的剂量下用药是足够安全的;为保障朱砂疗效的同时也保障其安全性,本发明对规定了hg含量的上下限,对本品中hg的下限按照实测平均值的80%规定为8398.39μg/g*80%*3g/袋=20.2mg/袋(此时转移率60%)制定;上限在比较朱砂按本品处方量的药典低限投料与药典规定hg允许日服量二者后,采用更严的前者制定为33.8mg/袋,因此最终本品hg限度范围为20.2mg~33.8mg/袋。
11、本发明对白芍、甘草、五味子、沉香的hplc含量测定,通过精密度试验,芍药苷色谱峰峰面积的平均值为5405.67,rsd为0.97%(n=6),沉香四醇色谱峰峰面积的平均值为1870.17,rsd为1.32%(n=6),甘草苷色谱峰峰面积的平均值为3119.67,rsd为0.82%(n=6),甘草酸色谱峰峰面积的平均值为1286.33,rsd为1.85%(n=6),五味子醇甲色谱峰峰面积的平均值为457.33,rsd为1.71%(n=6),表明所用仪器精密度良好;通过线性关系考察,5种成分在3.33-6811ng范围内线性关系良好;通过专属性试验,痰喘半夏颗粒中芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲5种成分均检出,而其各自的阴性样品中均未检出各自的指标色谱峰表明阴性对上述5种指标成分无干扰;通过稳定性试验,芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲5种成分的rsd分别为0.47%、071%、1.10%、1.0%、0.82%(n=6),表明样品溶液中5种成分在24h内稳定;通过重复性试验,6份样品中测得芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲的峰面积rsd分别为0.70%、0.68%、1.09%、0.96%、0.73%(n=6),表明该试验重复性良好;通过加样回收率试验,5种成分的平均加样回收率在90.83%~107.39%(药典规定1~10mg/g含量的回收率可接受范围是90%~108%;0.1~1mg/g含量回收率可接受范围是85%~110%),rsd0.88~1.83%(n=6)(药典规定1~10mg/g含量的rsd可接受范围是3%;0.1~1mg/g含量rsd可接受范围是4%),表明方法准确;通过耐用性试验,在不同品牌色谱柱上,5种成分含量的rsd小于3.0%,但在不同品牌仪器上,五味子醇甲含量(0.033mg/g)的rsd为8.5%(药典规定0.1~0.01mg/g含量的rsd可接受范围是11%),其他4种成分含量的rsd均小于3.4%,因此满足耐用性精密度要求。
附图说明:
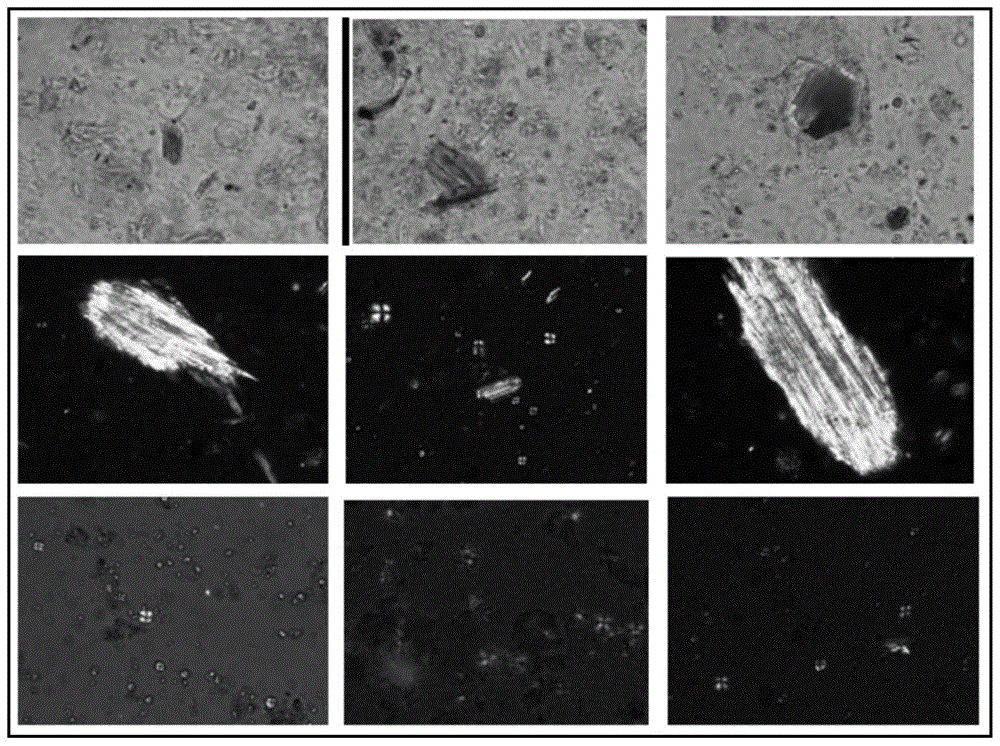
图1痰喘半夏颗粒中制半夏的草酸钙针晶束和淀粉粒显微图。
图2痰喘半夏颗粒中川贝母的淀粉粒显微图。
图3痰喘半夏颗粒中肉桂的石细胞和纤维显微图。
图4痰喘半夏颗粒中豆蔻的胚乳细胞淀粉团显微图。
图5痰喘半夏颗粒中沉香的导管显微图。
图6痰喘半夏颗粒中朱砂的碎片显微图。
图7痰喘半夏颗粒白芍薄层色谱图(从左到右:芍药苷对照品,样品(20170801),样品(20170102),样品(20170103),阴性,白芍对照药材)。
图8痰喘半夏颗粒白芍耐用性薄层色谱图(青岛普通预制g板)(从左到右:芍药苷对照品,样品(20170801)样品(20170102),样品(20170103),阴性,白芍对照药材)。
图9痰喘半夏颗粒白芍耐用性薄层色谱图(烟台普通预制g板)(从左到右:样品(20170801)样品(20170102),样品(20170103),阴性,白芍对照药材,芍药苷对照品)。
图10痰喘半夏颗粒白芍耐用性薄层色谱图(青岛高效g板)(从左到右:样品(20170801)样品(20170102),样品(20170103),阴性,白芍对照药材,芍药苷对照品)。
图11痰喘半夏颗粒白芍耐用性薄层色谱图(烟台高效g板)(从左到右:样品(20170801)样品(20170102),样品(20170103),阴性,白芍对照药材,芍药苷对照品)。
图12痰喘半夏颗粒白芍薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),样品(20170101),样品(20180302),样品(20160401),样品(20171202),样品(20171201),样品(20160602),样品(20160601),阴性,白芍对照药材,芍药苷对照品)。
图13痰喘半夏颗粒白芷对照品筛选薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,白芷对照药材,白芷素对照品)。
图14痰喘半夏颗粒白芷对照品筛选薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,白芷对照药材,欧前胡素对照品)。
图15痰喘半夏颗粒白芷点样量试验薄层色谱图(从左到右:样品(20170801)3μl,样品(20170801)5μl,样品(20170801)8μl,白芷对照药材5μl,阴性8μl)。
图16痰喘半夏颗粒白芷耐用性薄层色谱图(青岛高效g板)(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,白芷对照药材,欧前胡素对照品)。
图17痰喘半夏颗粒白芷耐用性薄层色谱图(烟台高效g板)(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,白芷对照药材,欧前胡素对照品)。
图18痰喘半夏颗粒白芷耐用性薄层色谱图(青岛普通预制g板)(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,白芷对照药材,欧前胡素对照品)。
图19痰喘半夏颗粒白芷耐用性薄层色谱图(烟台普通预制g板)(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,白芷对照药材,欧前胡素对照品)。
图20痰喘半夏颗粒白芷薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),样品(20170101),样品(20180302),样品(20160401),样品(20171202),样品(20171201),样品(20160602),样品(20160601),阴性,白芷对照药材,欧前胡素对照品)。
图21痰喘半夏颗粒五味子展开系统筛选薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,五味子对照药材,五味子醇甲对照品)。
图22痰喘半夏颗粒五味子展开系统筛选薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,五味子对照药材,五味子醇甲对照品)。
图23痰喘半夏颗粒五味子展开系统筛选薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,五味子对照药材,五味子醇甲对照品)。
图24痰喘半夏颗粒五味子展开系统筛选薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,五味子对照药材,五味子醇甲对照品)。
图25痰喘半夏颗粒五味子耐用性薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,五味子对照药材,五味子醇甲对照品)。
图26痰喘半夏颗粒五味子耐用性薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,五味子对照药材,五味子醇甲对照品)。
图27痰喘半夏颗粒五味子薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),样品(20170101),样品(20180302),样品(20160401),样品(20171202),样品(20171201),样品(20160602),样品(20160601),阴性,五味子对照药材,五味子醇甲对照品)。
图28痰喘半夏颗粒中酸枣仁、西洋参薄层色谱图(从左到右:缺西洋参阴性,西洋参对照药材,样品(20170801),样品(20170801)(无氨水洗涤步骤),酸枣仁对照药材,缺酸枣仁阴性,酸枣仁皂苷a对照品,酸枣仁皂苷b对照品)。
图29痰喘半夏颗粒酸枣仁前处理方法考察薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,酸枣仁对照药材,斯皮诺素对照品)。
图30痰喘半夏颗粒酸枣仁耐用性薄层色谱图(青岛普通预制g板)(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,酸枣仁对照药材,斯皮诺素对照品)。
图31痰喘半夏颗粒酸枣仁耐用性薄层色谱图(青岛高效g板)(从左到右:样品(20170801),样品(20170102),样品(20170103),阴性,酸枣仁对照药材,斯皮诺素对照品)。
图32痰喘半夏颗粒酸枣仁耐用性薄层色谱图(烟台高效g板)(从左到右:斯皮诺素对照品,样品(20170801),样品(20170102),样品(20170103),阴性,酸枣仁对照药材)。
图33痰喘半夏颗粒酸枣仁耐用性薄层色谱图(烟台普通预制g)(从左到右:样品(20170801),样品(20170102),样品(20170103),样品(20170103),阴性,酸枣仁对照药材,斯皮诺素对照品)。
图34痰喘半夏颗粒酸枣仁薄层色谱图(从左到右:样品(20170801),样品(20170102),样品(20170103),样品(20170101),样品(20180302),样品(20160401),样品(20171202),样品(20171201),样品(20160602),样品(20160601),阴性,酸枣仁对照药材,斯皮诺素对照品)。
图35痰喘半夏颗粒沉香液相鉴别图(250nm)(图35包括a-f图,1.沉香四醇,2.沉香特征峰)。
图36样品在不同波长的色谱图(图36包括a-e图)。
图37马兜铃酸a峰和干扰峰的uv光谱图。
图38马兜铃酸a的标准曲线图。
图39痰喘半夏颗粒中马兜铃酸a专属性hplc色谱图(图39包括a-d图)。
图40细辛药材色谱图。
图41细辛药材色谱图(a马兜铃酸a对照品、b供试品)。
图42不同提取时间考察结果折线图。
图43不同提取溶剂考察结果折线图。
图44不同料液比考察结果折线图。
图45芍药苷标准曲线图。
图46沉香四醇标准曲线图。
图47甘草苷标准曲线图。
图48甘草酸标准曲线图。
图49五味子醇甲标准曲线图。
图50痰喘半夏颗粒中5种指标成分专属性hplc色谱图(a.混合对照品溶液、b.供试品溶液、c.阴性溶液、ⅰ.缺白芍(235nm)、ⅱ.缺沉香(250nm)、ⅲ.缺甘草(①272nm、②250nm)、ⅳ.五味子(218nm)、1.芍药苷(235nm),2.沉香四醇(250nm),3.甘草苷(272nm),4.甘草酸(250nm),5.五味子醇甲(218nm))。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例1
【鉴别】(1)取本品研细,粉末用水溶解,过滤,取滤渣透化或直接用水装片置显微镜下观察,具体特征如下:
产品中制半夏的显微特征:淀粉粒圆形或类圆形,直径可至15~25μm,脐点多居中,呈短缝状、人字状,偏光下观察,脐点处汇聚的消光呈较对称的十字形;草酸钙针晶束,多断裂,长可至200~260μm;
产品中川贝母的显微特征:淀粉粒广卵形、长椭圆形、贝壳形、肾形,直径可至38~46μm,脐点点状、短缝状、人字状、星状,多位于较小端,偏光下观察,脐点处汇聚的消光常呈不对称的十字形,或呈л形和乂形;
产品中肉桂的显微特征:石细胞散在,多破碎,完整者呈类方形、类长方形、类圆形、圆多角形、椭圆形,直径32~60μm,壁厚,有的一面稍尖突,有的一面菲薄;纤维梭形,木化,边缘微波状,多断裂,长可至120~180μm,直径至40~60μm,璧极厚,孔沟不明显或稀少而细短,胞腔线形;
产品中豆蔻的显微特征:胚乳细胞淀粉团,长类方形、类圆形、不规则形,长可至120~180μm,由众多细小淀粉粒集结而成;
产品中沉香的显微特征:具缘纹孔导管碎片可见,具缘纹孔呈蜂窝状紧密排列;
产品中朱砂的显微特征:不规则颗粒和碎片散在,大小不一,鲜红色或暗红色,部分具光泽。
(2)取本品15g,研细,加乙醇40ml,超声处理20分钟,滤过,滤液蒸干,残渣加水15ml使溶解,用水饱和正丁醇萃取2次,每次15ml,合并正丁醇液,用氨水洗涤2次,每次15ml,弃去氨水,取正丁醇液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液,另取白芍对照药材0.5g,加乙醇15ml,同法制成对照药材溶液,再取芍药苷对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液,照通则0502薄层色谱法试验,吸取供试品溶液、对照药材溶液各15μl,对照品溶液10μl,分别点于同一硅胶g薄层板上,以比例为40:5:7:0.2的二氯甲烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至显色清晰,在日光下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
(3)取本品15g,研细,加乙醇20ml,超声处理15分钟,滤过,滤液蒸干,残渣加水15ml使溶解,用60℃石油醚萃取2次,每次20ml,合并石油醚液,蒸干,残渣加60℃石油醚1ml使溶解,作为供试品溶液,另取白芷对照药材1g,加乙醇15ml,同法制成对照药材溶液;再取欧前胡素对照品,加甲醇制成每1ml含0.04mg的溶液,作为对照品溶液;照通则0502薄层色谱法试验,吸取供试品溶液8μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以比例为10:5:0.2的环己烷-乙酸乙酯-甲醇为展开剂,展开,取出,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(4)取本品15g,加水40ml使溶解,离心5分钟,取上清液,置分液漏斗中,用水饱和正丁醇振摇提取2次,每次20ml,合并正丁醇液,用正丁醇饱和的水洗涤2次,每次20ml,取正丁醇液蒸干,残渣加甲醇1ml使溶解,置中性氧化铝柱(5g,100~200目,内径1cm,干法装柱)上,用甲醇20ml洗脱,收集洗脱液,浓缩至1ml作为供试品溶液;另取五味子对照药材1g,加甲醇10ml,超声处理10分钟,滤过,滤液浓缩至1ml,作为对照药材溶液;再取五味子醇甲对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液;照通则0502薄层色谱法试验,吸取供试品溶液5μl,对照药材溶液和对照品溶液各3μl,分别点于同一硅胶gf254薄层板上,以比例为12:4:0.4的甲苯-乙酸乙酯-冰醋酸为展开剂,展开,取出,晾干,在254nm紫外灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(5)取本品5g,研细,加甲醇40ml,超声处理20分钟,滤过,滤液蒸干,残渣加水3ml使溶解,置spec18小柱6cc上,依次用水3ml,35%的甲醇溶液3ml洗脱,弃去洗脱液,再用80%甲醇溶液3ml洗脱,收集洗脱液,洗脱液浓缩至1ml,作为供试品溶液;另取酸枣仁对照药材1g,加甲醇15ml,超声处理20分钟,滤过,滤液蒸干,加1ml甲醇使溶解,作为对照药材溶液,再取斯皮诺素对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液,照通则0502薄层色谱法试验,吸取供试品溶液15μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以水饱和正丁醇为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(6)取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入60%甲醇溶液15ml,称重,功率720w,频率40khz条件下超声处理20分钟,取出,再称重,用60%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;另取沉香对照药材0.5g,加60%甲醇溶液5ml,超声处理20分钟,滤过,取续滤液作为对照药材溶液,再取沉香四醇对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液,照通则0512高效液相色谱法试验,分别精密吸取供试品溶液、对照品溶液、对照品溶液各20μl,注入液相色谱仪,记录;供试品色谱图中,应呈现与沉香四醇对照品保留时间相一致的色谱峰,并同时呈现与对照药材中另一特征峰(其相对于沉香四醇的保留时间为2.95±1%)保留时间相一致的色谱峰;
高效液相色谱条件具体为:
采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长250nm,理论塔板数按芍药苷峰计算,应不低于15000;
【检查】马兜铃酸a照高效液相色谱法测定;
色谱条件以十八烷基硅烷键合硅胶为填充剂;以比例为38:62的乙腈~0.5%磷酸溶液为流动相;检测波长为270~350nm;理论塔板数按马兜铃酸a峰计算,应不低于30000;
对照品溶液的制备取马兜铃酸a对照品,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,摇匀,即得;
供试品溶液的制备取本品20袋,混匀,研细,取约20g,精密称定,置锥形瓶中,精密加入甲醇100~200ml,称重,摇匀,在功率720w,频率40khz条件下超声处理20~40分钟,取出,再称重,用甲醇补足减失重量,摇匀,滤过;精密吸取续滤液80~120ml,蒸干,残渣加0.5%氢氧化钠溶液20~40ml溶解,分次转移到分液漏斗中,用三氯甲烷萃取2~4次,每次20~30ml,弃去三氯甲烷液;水层用盐酸调ph至2,用三氯甲烷萃取2~5次,每次20~30ml,合并三氯甲烷液,蒸干,残渣用甲醇溶解并转移到10ml容量瓶中,加甲醇至刻度,摇匀,离心,取上清液,作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含马兜铃酸a(c17h11no7),不得过1.00μg;
重金属及有害元素照铅、镉、汞、砷、铜测定法(通则2321)测定,本品每袋含hg应为20.2~33.8mg。
【含量测定】白芍、甘草、沉香照通则0512高效液相色谱法测定;
色谱条件与系统适用性试验采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按规定进行梯度洗脱;芍药苷检测波长210nm、沉香四醇、甘草酸检测波长为210nm、甘草苷检测波长为215nm,理论塔板数按芍药苷峰计算,应不低于15000,所述梯度洗脱具体规定为:
对照品溶液的制备取芍药苷、沉香四醇、甘草苷、甘草酸对照品适量,加70%甲醇溶液制成每1ml含芍药苷200μg、沉香四醇40μg、甘草苷100μg和甘草酸80μg的混合溶液,摇匀,即得;
供试品溶液的制备取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入50%甲醇溶液5ml,称重,功率720w,频率40khz条件下超声处理30分钟,取出,再称重,用60%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含白芍以芍药苷(c23h28o11)计,不得少于4.3mg;含沉香以沉香四醇(c17h18o6)计,不得少于1.0mg;含甘草以甘草苷(c21h22o9)和甘草酸(c42h62o16)计,分别不得少于2.0mg和1.8mg。
实施例2
【鉴别】(1)取本品研细,粉末用水溶解,过滤,取滤渣透化或直接用水装片置显微镜下观察,具体特征如下:
产品中制半夏的显微特征:淀粉粒圆形或类圆形,直径可至15~25μm,脐点多居中,呈短缝状、人字状,偏光下观察,脐点处汇聚的消光呈较对称的十字形;草酸钙针晶束,多断裂,长可至200~260μm;
产品中川贝母的显微特征:淀粉粒广卵形、长椭圆形、贝壳形、肾形,直径可至38~46μm,脐点点状、短缝状、人字状、星状,多位于较小端,偏光下观察,脐点处汇聚的消光常呈不对称的十字形,或呈л形和乂形;
产品中肉桂的显微特征:石细胞散在,多破碎,完整者呈类方形、类长方形、类圆形、圆多角形、椭圆形,直径32~60μm,壁厚,有的一面稍尖突,有的一面菲薄;纤维梭形,木化,边缘微波状,多断裂,长可至120~180μm,直径至40~60μm,璧极厚,孔沟不明显或稀少而细短,胞腔线形;
产品中豆蔻的显微特征:胚乳细胞淀粉团,长类方形、类圆形、不规则形,长可至120~180μm,由众多细小淀粉粒集结而成;
产品中沉香的显微特征:具缘纹孔导管碎片可见,具缘纹孔呈蜂窝状紧密排列;
产品中朱砂的显微特征:不规则颗粒和碎片散在,大小不一,鲜红色或暗红色,部分具光泽。
(2)取本品25g,研细,加乙醇60ml,超声处理40分钟,滤过,滤液蒸干,残渣加水25ml使溶解,用水饱和正丁醇萃取4次,每次25ml,合并正丁醇液,用氨水洗涤4次,每次25ml,弃去氨水,取正丁醇液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液,另取白芍对照药材1.5g,加乙醇25ml,同法制成对照药材溶液,再取芍药苷对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液,照通则0502薄层色谱法试验,吸取供试品溶液、对照药材溶液各15μl,对照品溶液10μl,分别点于同一硅胶g薄层板上,以比例为40:5:7:0.2的二氯甲烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至显色清晰,在日光下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
(3)取本品25g,研细,加乙醇40ml,超声处理25分钟,滤过,滤液蒸干,残渣加水25ml使溶解,用90℃石油醚萃取4次,每次40ml,合并石油醚液,蒸干,残渣加90℃石油醚1ml使溶解,作为供试品溶液,另取白芷对照药材1g,加乙醇25ml,同法制成对照药材溶液;再取欧前胡素对照品,加甲醇制成每1ml含0.04mg的溶液,作为对照品溶液;照通则0502薄层色谱法试验,吸取供试品溶液8μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以比例为10:5:0.2的环己烷-乙酸乙酯-甲醇为展开剂,展开,取出,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(4)取本品25g,加水60ml使溶解,离心15分钟,取上清液,置分液漏斗中,用水饱和正丁醇振摇提取4次,每次40ml,合并正丁醇液,用正丁醇饱和的水洗涤4次,每次40ml,取正丁醇液蒸干,残渣加甲醇1ml使溶解,置中性氧化铝柱(5g,100~200目,内径1cm,干法装柱)上,用甲醇40ml洗脱,收集洗脱液,浓缩至1ml作为供试品溶液;另取五味子对照药材1g,加甲醇30ml,超声处理30分钟,滤过,滤液浓缩至1ml,作为对照药材溶液;再取五味子醇甲对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液;照通则0502薄层色谱法试验,吸取供试品溶液5μl,对照药材溶液和对照品溶液各3μl,分别点于同一硅胶gf254薄层板上,以比例为12:4:0.4的甲苯-乙酸乙酯-冰醋酸为展开剂,展开,取出,晾干,在254nm紫外灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(5)取本品15g,研细,加甲醇60ml,超声处理40分钟,滤过,滤液蒸干,残渣加水9ml使溶解,置spec18小柱(6cc)上,依次用水9ml,45%的甲醇溶液9ml洗脱,弃去洗脱液,再用90%甲醇溶液9ml洗脱,收集洗脱液,洗脱液浓缩至1ml,作为供试品溶液;另取酸枣仁对照药材1g,加甲醇25ml,超声处理40分钟,滤过,滤液蒸干,加1ml甲醇使溶解,作为对照药材溶液,再取斯皮诺素对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液,照通则0502薄层色谱法试验,吸取供试品溶液15μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以水饱和正丁醇为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(6)取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入80%甲醇溶液25ml,称重,功率720w,频率40khz条件下超声处理40分钟,取出,再称重,用80%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;另取沉香对照药材0.5g,加80%甲醇溶液15ml,超声处理40分钟,滤过,取续滤液作为对照药材溶液,再取沉香四醇对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液,照通则0512高效液相色谱法试验,分别精密吸取供试品溶液、对照品溶液、对照品溶液各20μl,注入液相色谱仪,记录;供试品色谱图中,应呈现与沉香四醇对照品保留时间相一致的色谱峰,并同时呈现与对照药材中另一特征峰(其相对于沉香四醇的保留时间为2.95±1%)保留时间相一致的色谱峰;
高效液相色谱条件具体为:
采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长250nm,理论塔板数按芍药苷峰计算,应不低于15000;
【检查】马兜铃酸a照高效液相色谱法测定;
色谱条件以十八烷基硅烷键合硅胶为填充剂;以比例为38:62的乙腈~0.5%磷酸溶液为流动相;检测波长为350nm;理论塔板数按马兜铃酸a峰计算,应不低于30000;
对照品溶液的制备取马兜铃酸a对照品,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,摇匀,即得;
供试品溶液的制备取本品20袋,混匀,研细,取约20g,精密称定,置锥形瓶中,精密加入甲醇200ml,称重,摇匀,在功率720w,频率40khz条件下超声处理40分钟,取出,再称重,用甲醇补足减失重量,摇匀,滤过;精密吸取续滤液120ml,蒸干,残渣加0.5%氢氧化钠溶液40ml溶解,分次转移到分液漏斗中,用三氯甲烷萃取4次,每次30ml,弃去三氯甲烷液;水层用盐酸调ph至2,用三氯甲烷萃取5次,每次30ml,合并三氯甲烷液,蒸干,残渣用甲醇溶解并转移到10ml容量瓶中,加甲醇至刻度,摇匀,离心,取上清液,作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含马兜铃酸a(c17h11no7),不得过1.00μg;
重金属及有害元素照铅、镉、汞、砷、铜测定法(通则2321)测定,本品每袋含hg应为20.2~33.8mg。
【含量测定】白芍、甘草、沉香照通则0512高效液相色谱法测定;
色谱条件与系统适用性试验采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按规定进行梯度洗脱;芍药苷检测波长255nm、沉香四醇、甘草酸检测波长为310nm、甘草苷检测波长为317nm,理论塔板数按芍药苷峰计算,应不低于15000,所述梯度洗脱具体规定为:
对照品溶液的制备取芍药苷、沉香四醇、甘草苷、甘草酸对照品适量,加70%甲醇溶液制成每1ml含芍药苷200μg、沉香四醇40μg、甘草苷100μg和甘草酸80μg的混合溶液,摇匀,即得;
供试品溶液的制备取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入90%甲醇溶液30ml,称重,功率720w,频率40khz条件下超声处理60分钟,取出,再称重,用80%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含白芍以芍药苷(c23h28o11)计,不得少于4.3mg;含沉香以沉香四醇(c17h18o6)计,不得少于1.0mg;含甘草以甘草苷(c21h22o9)和甘草酸(c42h62o16)计,分别不得少于2.0mg和1.8mg。
实施例3
【鉴别】(1)取本品研细,粉末用水溶解,过滤,取滤渣透化或直接用水装片置显微镜下观察;
淀粉粒圆形或类圆形,直径可至20μm,脐点多居中,呈短缝状、人字状,偏光下观察,脐点处汇聚的消光呈较对称的十字形;草酸钙针晶束,多断裂,长可至230μm;
淀粉粒广卵形、长椭圆形、贝壳形、肾形,直径可至43μm,脐点点状、短缝状、人字状、星状,多位于较小端,偏光下观察,脐点处汇聚的消光常呈不对称的十字形,或呈“л”形和“乂”形;
石细胞散在,多破碎,完整者呈类方形、类长方形、类圆形、圆多角形、椭圆形,直径32~60μm,壁厚,有的一面稍尖突,有的一面菲薄;纤维梭形,木化,边缘微波状,多断裂,长可至150μm,直径至50μm,璧极厚,孔沟不明显或稀少而细短,胞腔线形;
胚乳细胞淀粉团,长类方形、类圆形、不规则形,长可至150μm,由众多细小淀粉粒集结而成;
具缘纹孔导管碎片可见,具缘纹孔呈蜂窝状紧密排列;
不规则颗粒和碎片散在,大小不一,鲜红色或暗红色,部分具光泽;
(2)取本品20g,研细,加乙醇50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水20ml使溶解,用水饱和正丁醇萃取2次,每次20ml,合并正丁醇液,用氨水洗涤3次,每次20ml,弃去氨水,取正丁醇液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液;另取白芍对照药材1g,加乙醇20ml,同法制成对照药材溶液;再取芍药苷对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法试验,吸取供试品溶液、对照药材溶液各15μl,对照品溶液10μl,分别点于同一硅胶g薄层板上,以比例为40:5:7:0.2的二氯甲烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至显色清晰,在日光下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;
(3)取本品20g,研细,加乙醇30ml,超声处理20分钟,滤过,滤液蒸干,残渣加水20ml使溶解,用60℃~90℃石油醚萃取2次,每次30ml,合并石油醚液,蒸干,残渣加60℃~90℃石油醚1ml使溶解,作为供试品溶液;另取白芷对照药材1g,加乙醇20ml,同法制成对照药材溶液;再取欧前胡素对照品,加甲醇制成每1ml含0.04mg的溶液,作为对照品溶液;照薄层色谱法试验,吸取供试品溶液8μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以比例为10:5:0.2的环己烷-乙酸乙酯-甲醇为展开剂,展开,取出,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(4)取本品20g,加水50ml使溶解,离心10分钟,取上清液,置分液漏斗中,用水饱和正丁醇振摇提取2次,每次30ml,合并正丁醇液,用正丁醇饱和的水洗涤3次,每次30ml,取正丁醇液蒸干,残渣加甲醇1ml使溶解,置中性氧化铝柱上,用甲醇30ml洗脱,收集洗脱液,浓缩至1ml作为供试品溶液;另取五味子对照药材1g,加甲醇20ml,超声处理20分钟,滤过,滤液浓缩至1ml,作为对照药材溶液;再取五味子醇甲对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液;照通则0502薄层色谱法试验,吸取供试品溶液5μl,对照药材溶液和对照品溶液各3μl,分别点于同一硅胶gf254薄层板上,以比例为12:4:0.4的甲苯-乙酸乙酯-冰醋酸为展开剂,展开,取出,晾干,在254nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(5)取本品10g,研细,加甲醇50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水6ml使溶解,置spec18小柱(6cc)上,依次用水6ml,40%的甲醇溶液6ml洗脱,弃去洗脱液,再用90%甲醇溶液6ml洗脱,收集洗脱液,洗脱液浓缩至1ml,作为供试品溶液;另取酸枣仁对照药材1g,加甲醇20ml,超声处理30分钟,滤过,滤液蒸干,加1ml甲醇使溶解,作为对照药材溶液;再取斯皮诺素对照品,加甲醇制成每1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法试验,吸取供试品溶液15μl,对照药材溶液和对照品溶液各5μl,分别点于同一硅胶g薄层板上,以水饱和正丁醇为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,晾干,在365nm紫外灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(6)取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇溶液20ml,称重,功率720w,频率40khz条件下超声处理30分钟,取出,再称重,用70%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;另取沉香对照药材0.5g,加70%甲醇溶液10ml,超声处理30分钟,滤过,取续滤液作为对照药材溶液;再取沉香四醇对照品,加甲醇制成每1ml含0.1mg的溶液,作为对照品溶液;照高效液相色谱法试验,分别精密吸取供试品溶液、对照品溶液、对照品溶液各20μl,注入液相色谱仪,记录;供试品色谱图中,应呈现与沉香四醇对照品保留时间相一致的色谱峰,并同时呈现与对照药材中另一特征峰保留时间相一致的色谱峰;
所述高效液相色谱条件具体为:
采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按以下的规定进行梯度洗脱;检测波长250nm,理论塔板数按芍药苷峰计算,应不低于15000;
【检查】
马兜铃酸a照通则0512高效液相色谱法测定;
色谱条件以十八烷基硅烷键合硅胶为填充剂;以比例为38:62的乙腈~0.5%磷酸溶液为流动相;检测波长为320nm;理论塔板数按马兜铃酸a峰计算,应不低于30000;
对照品溶液的制备取马兜铃酸a对照品,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,摇匀,即得;
供试品溶液的制备取本品20袋,混匀,研细,取约20g,精密称定,置锥形瓶中,精密加入甲醇150ml,称重,摇匀,功率720w,频率40khz条件下超声处理30分钟,取出,再称重,用甲醇补足减失重量,摇匀,滤过;精密吸取续滤液100ml,蒸干,残渣加0.5%氢氧化钠溶液30ml溶解,分次转移到分液漏斗中,用三氯甲烷萃取3次,每次25ml,弃去三氯甲烷液;水层用盐酸调ph至2,用三氯甲烷萃取4次,每次25ml,合并三氯甲烷液,蒸干,残渣用甲醇溶解并转移到10ml容量瓶中,加甲醇至刻度,摇匀,离心,取上清液,作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含马兜铃酸a(c17h11no7),不得过1.00μg;
重金属及有害元素照通则2321铅、镉、汞、砷、铜测定法测定,本品每袋含hg应为20.2~33.8mg;
【含量测定】
(1)白芍、甘草、沉香照高效液相色谱法测定;
色谱条件与系统适用性试验采用二极管阵列检测器dad,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;芍药苷检测波长235nm、沉香四醇、甘草酸检测波长250nm、甘草苷检测波长272nm;理论塔板数按芍药苷峰计算,应不低于15000;
对照品溶液的制备取芍药苷、沉香四醇、甘草苷、甘草酸对照品适量,加70%甲醇溶液制成每1ml含芍药苷200μg、沉香四醇40μg、甘草苷100μg和甘草酸80μg的混合溶液,摇匀,即得;
供试品溶液的制备取本品10袋,混匀,研细,取约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇溶液20ml,称重,功率720w,频率40khz的条件超声处理30分钟,取出,再称重,用70%甲醇溶液补足减失重量,摇匀,滤过,取续滤液作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得;
本品每袋含白芍以芍药苷(c23h28o11)计,不得少于4.3mg;含沉香以沉香四醇(c17h18o6)计,不得少于1.0mg;含甘草以甘草苷(c21h22o9)和甘草酸(c42h62o16)计,分别不得少于2.0mg和1.8mg。
为了进一步验证本发明的有效性与科学性,发明人做了一系列的实验,具体如下:
实验例1:相对于痰喘半夏颗粒现行质量检测方法新增显微鉴别的验证
本品制法中制半夏、川贝母、肉桂、豆蔻(去壳)、沉香、丁香、西洋参、白芷、细辛、天竺黄、朱砂共11味药生粉入药,因此采用显微鉴别进行了观察,从中选取可视率较高的6味药(制半夏、川贝母、肉桂、豆蔻、沉香、朱砂)显微特征上标准正文显微鉴别项,见图1-6。采用德国来卡dm2500显微镜,摄像软件莱卡qwinv3。本品研细后粉末建议用水溶解,过滤后取滤渣透化或滤渣直接用水装片显微观察效果较佳,否则浸膏粉末制片背景颜色较深且模糊,不易找到显微特征。
本品中制半夏可见淀粉粒和大型草酸钙针晶束,其特点为淀粉粒脐点多居中,偏光下观察,脐点处汇聚的消光呈较对称的十字形;针晶较大型,成束和散在均可见,多断裂。川贝母淀粉粒具有特点,形状比半夏淀粉粒长而大,另可见贝壳形、肾形,脐点多位于较小端,偏光下观察,脐点处汇聚的消光常呈不对称的十字形,或呈“л”形和“乂”形。肉桂可见石细胞散在,破碎和完整者均可见,破碎者较多,细胞大而壁厚,有的一面稍尖突,有的一面菲薄;纤维大型、木化,多断裂,璧极厚,孔沟不明显或稀少而细短,胞腔线形。豆蔻可见胚乳细胞淀粉团,长类方形、类圆形、不规则形,较大,由众多细小淀粉粒集结而成。沉香可见具缘纹孔导管碎片可见,具缘纹孔呈蜂窝状紧密排列,在偏光下有荧光。朱砂可见不规则颗粒和碎片散在,大小不一,鲜红色或暗红色,部分具光泽,碎片在偏光下不具荧光现象。本品中淀粉粒众多,除制半夏和川贝母淀粉粒的形状和脐点有特点,易于区别外,其他药味中淀粉粒均有交叉,很难区分,因此没有列入其他药味淀粉粒。此外,草酸钙针晶束,除制半夏外,肉桂中也有,但肉桂针晶束较细小,与制半夏的大型针晶束易于区别。大型纤维除肉桂外,丁香中也有,但肉桂纤维木化,在偏光下有荧光,胞腔细长线形;丁香纤维微木化,胞腔宽窄不一。在没有上正文显微鉴别的药味中,丁香很难察见显微特征,花粉粒未见;丁香纤维偶见,但特征性不强。西洋参特征未察见,草酸钙簇晶未察见,淀粉粒无特征性。白芷淀粉粒特征不明显。细辛、天竺黄均未察见显微特征。
实验例2:相对于痰喘半夏颗粒现行质量检测方法新增白芍的薄层鉴别的验证
按方法对10批样品进行了检测和方法学验证。
芍药苷(批号110736-200732)、白芍对照药材(批号120905-201610)购自中检院。痰喘半夏颗粒样品10批及缺白芍阴性样品1批由贵州远程制药有限责任公司提供。试剂均为分析纯。
对照品溶液、对照药材溶液、供试品溶液制备同实施例3。
阴性样品制备同供试品溶液制备。
1方法:采用二氯甲烷-乙酸乙酯-甲醇-甲酸(40:5:7:0.2)为展开剂,对本品白芍鉴别进行试验。各溶液点样量同实施例3,展开约9cm,取出,晾干,置日光下观察。结果见图7。色谱图显示,以二氯甲烷-乙酸乙酯-甲醇-甲酸(40:5:7:0.2)为展开剂,适用本品中白芍的鉴别。供试品、对照药材、对照品、阴性样品均斑点清晰,且阴性无干扰。故确定该方法用于本品中白芍的薄层鉴别。
2方法学验证
2.1耐用性试验-不同厂家硅薄层板验证
采用4种不同薄层板,青岛高效g板、青岛普通预制g板、烟台高效g板、烟台普通预制g板进行验证。结果见图8-11,色谱效果重现性良好。
2.2专属性试验
取对照品溶液、供试品溶液、缺白芍的阴性溶液点于同一薄层板上。结果见图6-11,阴性溶液在芍药苷斑点相应位置上未出现斑点。说明专属性良好。
3样品检测
取10批痰喘半夏颗粒样品按实施例3的方法制备并检测,结果见图12,样品色谱图中均能检出芍药苷和白芍,10批样品均符合本发明规定。
实验例3相对于痰喘半夏颗粒现行质量检测方法新增白芷的薄层鉴别验证
按方法对10批样品进行了检测和方法学验证。
白芷对照药材(批号120945-201510)、白芷素对照品(批号110823-201606)、欧前胡素对照品(批号110826-201616)购自中检院。痰喘半夏颗粒样品10批及缺白芷阴性样品1批由贵州远程制药有限责任公司提供。试剂均为分析纯。
对照品溶液的制备取白芷素对照品,加甲醇制成每1ml含有1mg的白芷素对照品溶液;另取欧前胡素对照品加甲醇制成每1ml含有0.1mg的欧前胡素对照品溶液。
对照药材溶液、供试品溶液制备同实施例3。
阴性样品制备同供试品溶液制备。
1对照品的选择和点样量的试验
1.1白芷素对照品试验:
采用环己烷-乙酸乙酯-甲醇(1:1:0.2)对本品白芷中白芷素鉴别进行试验。白芷素对照品溶液点样量为1μl,白芷对照药材和供试品溶液点样量同实施例3,展开约9cm,置紫外365nm下检视,结果见图13。样品在白芷素对照品的相应位置上未出现相对应的荧光斑点。
1.2欧前胡素对照品试验
采用环己烷-乙酸乙酯-甲醇(10:5:0.2)为展开剂对本品白芷中欧前胡素鉴别进行试验,欧前胡素对照品溶液点样量为2μl,白芷对照药材和供试品点样量均同正文,展开约9cm,取出,晾干,置紫外光365nm下观察,结果见图14。样品在欧前胡素对照品和对照药材相应位置上均出现相对应的荧光斑点,且阴性无干扰,故采用此展开剂,并放弃白芷素对照品,以欧前胡素和白芷对照药材进行鉴别。
1.2不同点样量试验
取不同点样量的供试品、对照药材、阴性点样量进行试验。结果见图15,供试品为8μl,白芷对照药材5μl,阴性8μl时,主斑点的大小适中且清晰。故采用此点样量例入本发明检测方法中。
2方法学验证
2.1耐用性试验-不同厂家硅薄层板验证
采用4种不同薄层板,青岛高效g板、青岛普通预制g板、烟台高效g板、烟台普通预制g板进行验证。结果见图16-19,色谱效果重现性良好。
2.2专属性试验
取对照品溶液、供试品溶液、缺白芷的阴性溶液点于同一薄层板上。结果见图13-19,阴性溶液在欧前胡素斑点相应位置上未出现斑点,在白芷对照药材色谱图的相应位置上也未出现相应斑点。说明专属性良好。
3样品检测
取10批痰喘半夏颗粒样品按实施例3项下制备并检测,结果见图20,样品色谱图中均能检出欧前胡素和白芷,10批样品均符合本发明规定。
实验例4相对于痰喘半夏颗粒现行质量检测方法新增五味子的薄层鉴别验证
按方法对10批样品进行了检测和方法学验证。
五味子对照药材(批号120922-201309)、五味子醇甲(批号110857-201412)购自中检院。痰喘半夏颗粒样品10批及缺五味子阴性样品1批由贵州远程制药有限责任公司提供。试剂均为分析纯。
对照品溶液、对照药材溶液、供试品溶液制备同实施例3。
阴性样品制备同供试品溶液制备。
1展开剂的筛选
1.1选用展开剂甲苯-乙酸乙酯-冰醋酸(4:4:0.4)对本品五味子鉴别进行试验。各溶液点样量同正文,展开约9cm,取出,晾干,置紫外光254nm下观察,结果见图21。样品在对照品和对照药材的相应位置上有相对应的荧光斑点,但阴样品相应位置上有干扰,还需要继续优化。
1.2选用展开剂甲苯-乙酸乙酯-冰醋酸(8:4:0.4)对本品五味子鉴别进行试验。各溶液点样量同正文,展开约9cm,取出,晾干,置紫外光254nm下观察,结果见图22。样品在对照品和对照药材的相应位置上有相对应的荧光斑点,但阴样品相应位置上有干扰,还需要继续优化。
1.3选用展开剂甲苯-乙酸乙酯-冰醋酸(10:4:0.4)对本品五味子鉴别进行试验。各溶液点样量同正文,展开约9cm,取出,晾干,置紫外光254nm下观察,结果见图23。样品在对照品和对照药材的相应位置上有相对应的荧光斑点,但阴样品相应位置上有干扰,还需要继续优化。
1.4选用展开剂甲苯-乙酸乙酯-冰醋酸(12:4:0.4)对本品五味子鉴别进行试验。各溶液点样量同正文,展开约9cm,取出,晾干,置紫外光254nm下观察,结果见图24。样品在对照品和对照药材的相应位置上有相对应的荧光斑点,且阴样品相应位置上无干扰。
上述色谱图显示,以甲苯-乙酸乙酯-冰醋酸(12:4:0.4)为展开剂,适用本品中五味子的鉴别。供试品、对照品、对照药材、阴性样品均斑点清晰,且阴性无干扰。故确定该方法用于本品中五味子的薄层鉴别。
2方法学验证
2.1耐用性试验-不同厂家硅薄层板验证
采用2种不同薄层板,青岛gf254板、烟台gf254板进行验证。结果见图25、26,色谱效果重现性良好。
2.2专属性试验
取对照品溶液、供试品溶液、缺五味子的阴性溶液点于同一薄层板上。结果见图24-26,阴性溶液在五味子醇甲斑点相应位置上未出现斑点,说明专属性良好。3样品检测
取10批痰喘半夏颗粒样品按正文项下制备并检测,结果见图27,样品色谱图中均检出五味子醇甲和五味子,10批样品均符合本发明规定。
实验例5相对于痰喘半夏颗粒现行质量检测方法新增酸枣仁的薄层鉴别以及西洋参的薄层鉴别研究
按方法对10批样品进行了检测和方法学验证;并对3批样品进行了西洋参的薄层鉴别研究。
酸枣仁对照药材(批号121517-201604)、西洋参对照药材(批号120997-201309)、酸枣仁皂苷a(批号110734-201512)、酸枣仁皂苷b(批号110814-201408)、斯皮诺素对照品(批号111869-201203)购自中检院。痰喘半夏颗粒样品10批及缺酸枣仁阴性、缺西洋参阴性样品各1批由贵州远程制药有限责任公司提供。试剂均为分析纯。
1皂苷类成分的薄层鉴别
1.1对照品溶液的制备
取酸枣仁皂苷a、酸枣仁皂苷b、人参皂苷f11对照品适量,加甲醇制成每1ml含酸枣仁皂苷a、酸枣仁皂苷b、人参皂苷f11各1mg的对照品溶液。
1.2对照药材溶液的制备
取酸枣仁对照药材2g,加甲醇30ml,加热回流60min,滤过,滤液浓缩至5ml作为酸枣仁对照药材溶液。
再取西洋参对照药材2g,加甲醇30ml,超声处理30min,滤过,滤液浓缩至5ml作为西洋参对照药材溶液。
1.3供试品溶液的制备
取样品平行制备2份,每份20g,研细,加甲醇100ml,加热回流60min,滤过,滤液蒸干,残渣加水20ml使溶解,加入水饱和正丁醇萃取2次,每次20ml,合并正丁醇液(备用),另一份用氨水洗涤3次,每次20ml,弃去氨水,取上述二份正丁醇液加入正丁醇饱和的水洗涤3次,每次20ml,弃去正丁醇饱和的水,取正丁醇液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。另再取缺酸枣仁的阴性对照药材20g,同供试品方法法制成阴性对照药材溶液。
1.4阴性溶液的制备
取缺酸枣仁和缺西洋参的阴性样品各20g,同供试品制备法(未经氨水洗涤)分别制成各自的阴性溶液。
1.5方法筛选和结果
1.5.1参考文献中展开剂,照薄层色谱法(中国药典通则0502)试验,吸取两种供试品溶液各10μl,两种对照药材溶液各5μl,两种阴性溶液各10μl,酸枣仁皂苷a、酸枣仁皂苷b对照品溶液各5μl,分别点于同一硅胶g薄层板上,以正丁醇-冰醋酸-水(4:1:5)的上层溶液为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,于105℃加热至显色清晰,置于日光下检视。
结果见图28。供试品溶液、酸枣仁对照药材和西洋参对照药材、2种阴性的色谱图在酸枣仁皂苷a、b对照品相应位置上的斑点基本没有区别,因此所选择的展开剂不适用于酸枣仁皂苷与西洋参皂苷的区分。
2黄酮类成分的薄层鉴别
2.1对照品溶液的制备
取斯皮诺素对照品,制备方法同实施例3。
2.2对照药材溶液制备
制备方法同实施例3。
2.3供试品溶液制备
同实施例3。采用spec18小柱(6cc)净化方法制备酸枣仁供试品溶液。
2.4阴性样品制备
同供试品溶液制备。
2.5方法和结果
以水饱和正丁醇为展开剂,取供试品溶液、对照药材溶液、阴性溶液点样,展开,取出,晾干,喷以1%香草醛硫酸溶液,在紫外灯365nm下检视。结果见图29,样品在斯皮诺素对照品和酸枣仁对照药材的相应位置上均出现相应的荧光斑点,故采用此方法作为本品中酸枣仁的薄层鉴别方法。
2方法学验证
2.1耐用性试验-不同厂家硅薄层板验证
采用4种不同薄层板,青岛高效g板、青岛普通预制g板、烟台高效g板、烟台普通预制g板进行验证。结果见图30-33,色谱效果重现性良好。
2.2专属性试验
取对照品溶液、供试品溶液、缺酸枣仁的阴性溶液点于同一薄层板上。结果见图28-33,阴性溶液在斯皮诺素斑点相应位置上未出现相应斑点,在酸枣仁对照药材溶液的相应位置上也未出现相应斑点。说明专属性良好。
3样品检测
取10批痰喘半夏颗粒样品按正文项下制备并检测,结果见图34,样品色谱图中均检出斯皮诺素和酸枣仁,10批样品均符合本发明的规定。
实验例6相对于痰喘半夏颗粒现行质量检测方法新增沉香的液相鉴别
照高效液相色谱法(通则0512)进行鉴别。取10批样品按照实施例3制备并进行检测。
沉香四醇(批号:111980-201602,纯度≥98.3%)、沉香对照药材(批号120922-201309)购自中检院。痰喘半夏颗粒样品10批及缺沉香阴性样品由贵州远程制药有限责任公司提供。试剂均为分析纯。仪器为agilent1260高效液相仪(美国安捷伦公司)。
1色谱条件与系统适用性试验同实施例3。
2对照品溶液的制备取沉香四醇对照品按实施例3制备。
3供试品溶液的制备同实施例3。
4阴性样品制备同供试品溶液制备。
5方法和结果
取对照品溶液、对照药材溶液、供试品溶液、阴性样品各20μl,分别注入色谱仪,以【含量测定】项下色谱条件检测。结果见图35,沉香对照药材色谱图中,沉香四醇的保留时间为18.020min,以沉香四醇为参照(s),保留时间为54.410的沉香特征峰的相对保留时间(rrt)为54.410/18.020=3.0194。供试品溶液色谱图中检出了沉香四醇和沉香特征峰,且阴性未出现该二个峰,表明无干扰。因此该方法适合于本品中沉香药材的鉴别。
6样品检测
取10批痰喘半夏颗粒样品按正文项下制备并检测,结果样品色谱图中均检出沉香四醇和相对平均保留时间为2.95的沉香特征峰,10批样品中沉香特征峰的rrt计算见表1,特征峰相对保留时间(rrt)均值为2.9509。10批样品均符合本发明的规定。
表110批样品中沉香特征峰的rrt计算
实验例7检查项验证
【检查】(一)水分限度维持原标准,不得过5.0%。药典对颗粒剂水分除另有规定外,不得过8.0%。本品原标准要求较药典严,不做修订。
(二)马兜铃酸a的限量检查照高效液相色谱法(通则0512)测定。
仪器agilent1260高效液相色谱仪(美国安捷伦公司),js-720型超声仪(昆山禾创超声设备有限公司),merckmillipore超纯水制备系统(德国默克公司)。
试药马兜铃酸a(批号:110746-201611纯度≥96.5%)购自中国食品药品检定研究院。乙腈、磷酸为色谱纯,水为超纯水,其他试剂均为分析纯。共收集到1家企业(贵州远程制药有限责任公司)的10批样品。缺细辛的阴性样品、细辛药材均由贵州远程制药有限责任公司提供。
1色谱条件与系统适用性试验同实施例3。
色谱方法参考药典细辛中马兜铃酸a的检查方法。检测波长的选择:马兜铃酸a在225、249、260、320nm处有较大吸收,药典选择260nm,但在本品中有一小杂峰在马豆玲酸a峰位置出现且难以分开,见图36、37,但该杂峰在360nm检测时可以消除干扰,故选择没有干扰的320nm,此处马豆玲酸a峰并不是最高吸收,其峰高虽然有所下降,但依然能保证方法灵敏度。
2溶液的制备
2.1对照品溶液的制备取马兜铃酸a对照品同实施例3的方法配制。
2.2供试品溶液的制备配制同实施例3的方法配制。
样品前处理方法其原理为:马兜铃酸a能溶于甲醇等有机溶剂,也溶于碱液。提取液经碱化后用三氯甲烷除杂,再经酸化后三氯甲烷萃取,可得到较好的净化和浓缩效果,是马兜铃酸a痕量测定的经典方法。本研究比较了不同浓度甲醇超声提取结果,确定了第一步采用甲醇提取,再用上述经典方法除杂,可克服痕量的马兜铃酸a在痰喘半夏颗粒复杂背景中难以检测的困难,该提取方法的准确性通过后面的加样回收得到了证明。
2.3阴性样品溶液的制备取缺细辛的阴性样品,按供试品项下方法制备。
2.4细辛药材溶液的制备按供试品溶液制备方法制备。
3方法学考察
3.1精密度试验
精密吸取马兜铃酸a对照品溶液20μl,注入液相色谱仪,连续进样6次。结果见表2,马兜铃酸a色谱峰峰面积的平均值为4457,rsd为0.08%(n=6),表明仪器精密度良好。
表2马兜铃酸a的精密度实验结果
3.2线性关系考察
分别精密吸取马兜铃酸a对照品溶液1、5、10、15、20、30μl,注入液相色谱仪,结果见表3,以进样量(μg)为横坐标,峰面积(a)为纵坐标,绘制标准曲线,见图38,回归方程为y=2452x-6.111,r=0.99999(n=6),马兜铃a在0.1239~3.717μg范围内线性关系良好。
表3马兜铃酸a的线性关系考察结果
3.3专属性试验
取对照品溶液、供试品溶液和阴性样品溶液各20μl,分别注入液相色谱仪测定,结果见图39。缺细辛的阴性样品在马兜铃酸a相应保留时间区域未出现色谱峰,表明阴性样品对供试品检测无干扰。由于供试品溶液中未检出马兜铃酸a色谱峰,所以取加样回收率项下溶液进样,加标样品中检出马兜铃酸a峰。
3.4稳定性试验
精密吸取同一份加样回收率试验项下溶液(批号:批号20170103)20μl,在该色谱条件下于0、2、6、8、16、24h分别进样。马兜铃酸a的峰面积值的平均值3866,rsd为0.53%(n=6),见表4,表明上述样品溶液在24h内稳定。
表4痰喘半夏颗粒中马兜铃酸a稳定性实验结果
3.5重复性试验
取同一批痰喘半夏颗粒(批号20170103)6份,每份20g,精密称定,按照供试品溶液制备项下制备,精密吸取20μl,注入液相色谱仪中测定,6份样品中均未检测到马兜铃酸a峰。
3.6加样回收率试验
取同一批号样品(批号20170103)6份,每份10g,精密称定,精密加入马兜铃酸a对照品约1.5mg(在本品中含量为0.015%,相当于细辛中含量为0.14%)。按供试品溶液制备方法制备,精密吸取20μl,注入液相色谱仪,测定,计算加样回收率。结果见表5,马兜铃酸a的回收率在83.68%~87.69%,平均回收率85.40%(药典规定0.01%含量的回收率应在85%-110%),rsd为1.55%(n=6)。
表5痰喘半夏颗粒中马兜铃酸a加样回收率实验结果
3.7检测限试验
精密吸取马兜铃酸a对照品溶液1ml(浓度为0.1031mg·ml-1),置100ml容量瓶中,加甲醇稀释至刻度,摇匀,再精密吸取此溶液1ml,置20ml中,加甲醇稀释至刻度,摇匀,精密吸取10μl,按以上色谱条件测定,计算信噪比(s/n),此时s/n=3.4,因此马兜铃酸a的检测限=0.1031μg/μl*1/100*1/20*10μl*1000=0.5155ng。
3.8耐用性试验
采用重复性项下供试品(批号20170103),分别在3根不同品牌的c18色谱柱上进样:ymchydrosphere-c18色谱柱(250mm×4.6mm,5μm);agela-durashellrp色谱柱(250mm×4.6mm,5μm);welch-topsilc18色谱柱(250mm×4.6mm,5μm)。再在3台不同品牌的高效液相色谱仪上进样。结果见表6,供试品中均未检出马兜铃酸a。
表6痰喘半夏颗粒中马兜铃酸ahplc检查耐用性试验结果
3.9理论塔板数的确定
计算加样回收率项下马兜铃酸a峰的理论塔板数和分离度,见表7,根据平均值确定本品马兜铃酸a峰的理论塔板数应不低于30000。
表7马兜铃酸a理论板数和分离度统计表
4样品检测
4.1痰喘半夏颗粒中马兜铃酸a的检测
取上述10批痰喘半夏颗粒,按供试品溶液制备方法制备,精密吸取20μl,分别注入液相色谱仪中测定,按外标一点法计算马兜铃酸a的含量,结果见表8,10批样品中均未检出马兜铃酸a。
表8痰喘半夏颗粒中马兜铃酸a检测结果
4.2细辛药材中马兜铃酸a的检测
4.2.1按痰喘半夏颗粒提取方法
取处方中的细辛药材(20170103批次痰喘半夏颗粒投料,厂方提供),粉碎,取粉末2g,精密称定,按痰喘半夏颗粒供试品溶液制备方法制备,精密吸取20μl,注入液相色谱仪,依法测定,结果如图40,同样也未检出马兜铃酸a。
4.2.2按药典细辛项下方法
取处方中的细辛药材(20170103批次痰喘半夏颗粒投料,厂方提供),按2015版药典细辛项下方法测定:以ymchydrosphere-c18(250mm×4.6mm,5μm)色谱柱,乙腈为流动相a,0.05%磷酸溶液为流动相b,梯度洗脱(0~10min,30→34%a;10~18min,34→35%a;18~20min,35→45%a;20~30min,45%a;30~31min,45→53%a;31~35min,53%a;35~40min,53→100%a);检测波长260。理论塔板数按马兜铃酸a峰计算不低于5000。取马兜铃酸a对照品适量,精密称定,加甲醇配制成每1ml含0.230μg的对照品溶液。取细辛药材粉碎,取粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率720w,频率40khz)40分钟,放冷,再称定重量,用70%甲醇补足减失重量,摇匀,滤过,取续滤液,作为供试品溶液。精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,结果见图41,在细辛药材中同样也未检出马兜铃酸a。
5痰喘半夏颗粒中马兜铃酸a限度的制定
如果用符合药典规定(≤0.001%)的细辛投料,则本品中马兜铃酸a含量=0.001%*107/1000=1.07μg/g(相当于3.21μg/袋),此时供试品溶液进样的马兜铃酸a量=1.07μg/g*20g/150ml*100ml/10ml*20μl=28.5ng,马兜铃酸a的仪器检测限为0.5155ng,因此样品中如果用药典低限的细辛投料则所含马兜铃酸a量是检测限的55倍,完全满足检测要求。根据药典中细辛日服量3g,得到马兜铃酸a的日服量为0.001%*3g=30μg,痰喘半夏颗粒日服量为9g,则马兜铃酸a限度=30μg/9g=0.33μg/g(1.00μg/袋),此时是检测限的18倍,也完全满足检测要求。最终按照更严的日服量来制定痰喘半夏颗粒中马兜铃酸a限量为不得过1.00μg/袋。
重金属及有害元素照重金属检查法(通则2321)中的电感耦合等离子质谱法检测。
仪器agilent7700电感耦合等离子体质谱仪(美国安捷伦公司),mars6classic微波消解仪(美国培安科技公司),agilent1260高效液相仪(美国安捷伦公司),js-720型超声仪(昆山禾创超声设备有限公司),merckmillipore超纯水制备系统(德国默克公司)。
试药铅元素标准溶液(标准物质号:gbw08619规格:1000μg/ml)、镉元素标准溶液(标准物质号:gbw08612规格:1000μg/ml)、砷元素标准溶液(标准物质号:gbw08611规格:1000μg/ml)、汞元素标准溶液(标准物质号:gbw08617规格:1000μg/ml)、铜元素标准溶液(标准物质号:gbw08615规格:1000μg/ml)均购自中国计量科学研究院。锗元素标准溶液(标准物质号:gsb04-1728-2004规格:1000μg/ml)、铋元素标准溶液(标准物质号:gsb04-1719-2004规格:1000μg/ml)、铟元素标准溶液(标准物质号:gsb04-1731-2004规格:1000μg/ml)购自国家有色金属及电子分析测试中心。高纯氩气(南京天泽气体,纯度>99.999%),硝酸(mos级,南京化学试剂有限公司),水为去离子水,其他试剂均为分析纯。
1质谱条件
冷却气流量15l/min;辅助气流量0.8l/min;载气0.8l/min;补偿气流量0.8l/min;雾化器babington;扫描次数3次;采集模式spectrum;检测方式自动。
2标准曲线溶液的制备
精密吸称取铅(pb)、镉(cd)、砷(as)、铜(cu)4种元素的标准溶液适量,加入10%硝酸配制成含pb1、5、10、20ng/ml,含cd0.5、2.5、5、10ng/ml,含as1、5、10、20ng/ml,含cu50、100、200、500ng/ml的混合标准曲线溶液。精密吸称取汞(hg)的标准溶液适量,加入10%硝酸配制成含hg0.2、0.5、1、2、5ng/ml的单元素标准曲线溶液。
3内标溶液的制备
精密吸称取锗(ge)、铋(bi)、铟(in)的标准溶液适量,加入10%硝酸配制成含3种元素各1000ng/ml的混合内标溶液。
4供试品溶液的制备
取本品10袋,混匀,取约0.1g,研细,精密称定,置聚四氟乙烯微波消解罐中,加入硝酸5ml,密塞,拧紧盖子,装上保护套,按表9方法消解,消解后放冷,将消解液用去离子水分次洗涤、转移到50ml容量瓶中,并定容至刻度,摇匀,再精密吸取1ml于25ml的容量瓶中,用去离子水稀释至刻度,摇匀,作为供试品溶液①。另精密吸取取供试品溶液①1ml,置于250ml容量瓶中,用去离子水稀释至刻度,摇匀,作为供试品溶液②。
表9微波消解程序表
5空白对照溶液制备
同供试品方法平行制备,作为测定随行的空白对照。
6方法学考察
6.1线性关系、检测限试验
根据痰喘半夏颗粒中hg含量以及其他4种元素的含量制定合适的线性范围,方法检测限根据线性自动计算得出,结果见表10。各元素在各自的浓度范围内线性关系良好(r≥0.9968),5个元素的检测限在0.0200-0.0793ng/ml。
表10方法检出限、线性范围和相关系数结果
6.2朱砂专属性试验
取不含朱砂的阴性样品,同供试品溶液制备方法制备,依上法检测。结果,pb、cd、hg、cu的含量分别为0.03、0.03、0.73、5.61μg/g,as未检出。
7样品测定
取10批样品,按供试品溶液制备方法分别制备供试品溶液①和②,分别进样,进样前以空白对照溶液进行校正,结果见表11。10批样品中各元素平均含量为:pb0.03μg/g(1批未检出);cd0.02μg/g;as在10批样品中均未检出;hg8398.39μg/g;cu5.03μg/g(1批未检出)。
表11痰喘半夏颗粒中5种重金属及有害元素和测定结果
8重金属及有害元素限度的制定
结合10批样品的测定结果,参照2015版《中国药典》中甘草项下的相关规定:pb≤5.0mg/kg、cd≤0.3mg/kg、as≤2.0mg/kg、cu≤20.0mg/kg、hg≤0.2mg/kg。根据上述结果,所有批次中的pb、cd、as、cu的含量均远低于该标准要求,因此此4个元素不上质量检测方法正文。
朱砂中含有的hg既是有效成分,又是毒性成分。测定结果表明hg在痰喘半夏颗粒中含量很高,由阴性可知,主要是来自朱砂。本品朱砂以水飞后的极细粉入药,药典规定水飞朱砂hgs含量不得少于98.0%,相当于hg为84.5%;因此本品中hg含量应不得少于84.5%*13.333g/1000g=11266.67μg/g。如按样品中hg实测均值8398.39μg/g计算,则朱砂转移率为74.5%;如按实测最低值6142.80μg/g计算,则朱砂转移率为54.5%,显然在本品中转移率偏低,有可能水飞时损失严重。
朱砂有毒,不宜大量服用,在药典中的最大日服量为0.5g,因此以朱砂中hg含量84.5%计算则hg的日服量不得过0.43g。本品最大日服量为9g,如果本品严格按照朱砂处方量投料,则本品中hg的日服量为84.5%*13.333g/1000g*9g=0.1014g/天(相当于本品中hg为33.8mg/袋),远低于药典规定的0.43g/天(相当于本品中hg为0.143g/袋)。表明本品中朱砂的处方量以及在规定的剂量下用药是足够安全的。
为保障朱砂疗效的同时也保障其安全性,应对其规定hg含量的上下限。故对本品中hg的下限按照实测平均值的80%规定为8398.39μg/g*80%*3g/袋=20.2mg/袋(此时转移率60%)制定;上限在比较朱砂按本品处方量的药典低限投料与药典规定hg允许日服量二者后,采用更严的前者制定为33.8mg/袋。。因此最终本品hg限度范围为20.2mg~33.8mg/袋。
实验例8白芍、甘草、五味子、沉香的hplc含量测定
仪器agilent1260高效液相色谱仪(美国安捷伦公司),js-720型超声仪(昆山禾创超声设备有限公司),merckmillipore超纯水制备系统(德国默克公司)。
试药芍药苷(批号:110736-201035纯度≥96.5%)、沉香四醇(批号:111980-201602,纯度≥98.3%)、甘草苷(批号:111610-201607,纯度≥93.1%)、甘草酸(批号:110731-201619,纯度≥93.0%)、五味子醇甲(批号:110855-201311,纯度≥98.1%)均购自中国食品药品检定研究院。购自中国食品药品检定研究院。乙腈、磷酸为色谱纯,水为超纯水,其他试剂均为分析纯。
样品共收集到1家企业(贵州远程制药有限责任公司)的10批样品,样品批号分别为20160401、20160601、20160602、20170101、20170102、20170103、20170801、20171201、20171202、20180302。缺白芍阴性样品、缺甘草阴性样品、缺沉香阴性样品、缺五味子阴性样品均由贵州远程制药有限责任公司提供。
1色谱条件
以ymc-hydrospherec18色谱柱(250mm×4.6mm,5μm)为色谱柱,以乙腈-0.5%磷酸溶液为流动相,梯度洗脱,0min15%a;24min18%a;28min38%a;59min47%a。流速1.0ml·min-1,柱温30℃。检测波长的选择:取混合对照品溶液,在200nm-400nm波长范围内提取光谱,显示沉香四醇在215、275、310nm处有较大吸收;甘草苷在215、235、272、317nm处有较大吸收;甘草酸在250nm处为最大吸收波长;五味子醇甲在218、249nm处有较大吸收。根据其uv光谱信息,采用统一检测波长的可能性在218nm,235nm,250nm或272nm,但考察215nm时,甘草苷、甘草酸均有杂质干扰;考察235nm时,甘草酸有干扰,且五味子峰面积太小。考察250nm时,芍药苷、五味子醇甲的峰面积太小。考察272nm时,芍药苷、五味子醇甲、沉香四醇峰面积太小。故最终采用二极管阵列检测器选择各自的波长测定,选取218nm为五味子醇甲的检测波长、235nm芍为药苷的检测波长,250nm为沉香四醇、甘草酸的检测波长,272nm为甘草苷的检测波长,能实现5种成分的同时测定并消除干扰。
2溶液的制备
2.1混合对照品溶液的制备
精密称取芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲对照品适量,精密称定,加70%甲醇制成每1.0ml含芍药苷195.51μg、沉香四醇42.80μg、甘草苷91.31μg、甘草酸77.60μg、五味子醇甲3.96μg的混合对照品溶液,4℃保存备用。
2.2供试品溶液的制备同实施例3。
2.3阴性样品溶液的制备
称取缺白芍阴性样品、缺甘草阴性样品、缺沉香阴性样品、缺五味子阴性样品各约2g,精密称定,按供试品方法分别制备阴性样品溶液。
3样品前处理方法考察
3.1提取方法考察
取本品粉末约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇10ml,密塞,称定重量,分别采用超声处理(功率720w,频率40khz)30min和加热回流30min的提取方式提取,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,离心,取上清液,进样,结果见表12。
表12不同提取方法考察结果
结果表明:超声与回流两种方法提取效果差异较大,超声提取效果较好,故选择超声得方法来处理样品。
5.2提取时间考察
取本品粉末约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇10ml,密塞,称定重量,采用超声处理(功率720w,频率40khz)30、45、60min分别提取,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,离心,取上清液,进样,结果见表13和图42。
表13不同提取时间考察结果
结果表明:芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲的峰面积随提取时间增加无显著变化,故选用30min的超声提取时间。
5.3不同提取溶剂考察
取本品粉末约2g,精密称定,置具塞锥形瓶中,精密加入50%甲醇、70%甲醇、90%甲醇、甲醇各10ml,密塞,称定重量,采用超声处理(功率720w,频率40khz)30min,取出,放冷,再称定重量,分别用上述溶剂各自补足减失的重量,摇匀,离心,取上清液,进样。结果见表14和图43。
表14不同提取溶剂考察结果
结果表明:芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲的峰面积在70%甲醇的提取溶剂下峰面积最大,故选用70%甲醇作为提取溶剂。
5.4不同料液比考察
取本品粉末约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇5、10、15、20、30ml,密塞,称定重量,超声处理(功率720w,频率40khz)30min,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,离心,取上清液,进样,结果见表15和图44。
表15不同料液比考察结果
结果表明:芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲的峰面积随溶剂量增加而无显著差异,考虑到五味子醇甲的峰面积不宜过小,综合考虑,选择20ml作为最佳溶剂量,此时料液比为10/1(ml/g)。
5.5前处理方法的确定
最终确定前处理方法为:取本品粉末2g,加70%甲醇20ml,超声处理(功率720w,频率40khz)30min,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,离心,取上清液作为供试品溶液。
6方法学考察
6.1精密度试验
精密吸取混合对照品溶液20μl,注入液相色谱仪中,连续进样6次,记录各检测波长下目标色谱峰的面积。结果见表16,芍药苷色谱峰峰面积的平均值为5405.67,rsd为0.97%(n=6);沉香四醇色谱峰峰面积的平均值为1870.17,rsd为1.32%(n=6);甘草苷色谱峰峰面积的平均值为3119.67,rsd为0.82%(n=6);甘草酸色谱峰峰面积的平均值为1286.33,rsd为1.85%(n=6);五味子醇甲色谱峰峰面积的平均值为457.33,rsd为1.71%(n=6);表明所用仪器精密度良好。
表16痰喘半夏颗粒中5种指标成分的精密度实验结果
6.2线性关系考察
取混合对照品溶液,分别精密吸取1、3、9、15、25、35μl,进样,记录。以混合对照品的进样量为横坐标,以峰面积值为纵坐标,绘制标准曲线,结果见图45-49、表17、18,5种成分在3.33-6811ng范围内线性关系良好。
表17痰喘半夏颗粒中5种指标成分的线性关系考察结果
表18痰喘半夏颗粒中5种指标成分的线性回归方程及线性范围
6.3专属性试验
取对照品溶液、供试品溶液和阴性样品溶液各20μl,分别注入液相色谱仪,记录,结果见图50。痰喘半夏颗粒中芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲5种成分均检出,而其各自的阴性样品中均未检出各自的指标色谱峰表明阴性对上述5种指标成分无干扰。
6.4稳定性试验
取样品(批号20170103)1份,按供试品溶液制备方法制备,于0、4、8、12、16、20、24h分别进样,结果见表21。芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲5种成分的rsd分别为0.47%、071%、1.10%、1.0%、0.82%(n=6),表明样品溶液中5种成分在24h内稳定。
表21痰喘半夏颗粒中5种指标成分的稳定性实验结果
6.5重复性试验
取同一批痰喘半夏颗粒(批号:批号20170103)6份,每份约2g,精密称定,按照供试品溶液制备方法平行制备,精密吸取20μl,注入液相色谱仪中,结果见表19。6份样品中测得芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲的峰面积rsd分别为0.70%、0.68%、1.09%、0.96%、0.73%(n=6),表明该试验重复性良好。
表19痰喘半夏颗粒中5种指标成分的重复性实验结果
6.6加样回收率试验
取同一批号样品(批号20170103)6份,每份约1g,精密称定,置具塞锥形瓶中,精密加入混合对照品溶液10ml,按供试品方法制备。精密吸取20μl,注入液相色谱仪,测定。计算加样回收率,结果见表20。5种成分的平均加样回收率在90.83%~107.39%(药典规定1~10mg/g含量的回收率可接受范围是90%~108%;0.1~1mg/g含量回收率可接受范围是85%~110%),rsd0.88~1.83%(n=6)(药典规定1~10mg/g含量的rsd可接受范围是3%;0.1~1mg/g含量rsd可接受范围是4%),表明方法准确。
表20痰喘半夏颗粒中5种指标成分的加样回收率实验结果
6.7耐用性试验
取样品(批号20160401)1份,按供试品溶液制备方法制备,于不同品牌色谱柱和仪器下分别进样。结果见表22、23,在不同品牌色谱柱上,5种成分含量的rsd小于3.0%;但在不同品牌仪器上,五味子醇甲含量(0.033mg/g)的rsd为8.5%(药典规定0.1~0.01mg/g含量的rsd可接受范围是11%),其他4种成分含量的rsd均小于3.4%,因此满足耐用性精密度要求。
表22痰喘半夏颗粒中5种指标成分的耐用性试验-不同色谱柱(aglient1260仪器)
表23痰喘半夏颗粒中5种指标成分的耐用性试验-不同液相仪(ymchydrosphere-c18)
6.8理论塔板数的确定
统计不同批次样品中5种成分的理论塔板数和分离度,结果见表34,芍药苷的理论塔板数大于18000,分离度大于1.4;沉香四醇的理论塔板数大于27000,分离度大于1.8;甘草苷的理论塔板数大于11000,分离度大于1.2;甘草酸的理论塔板数大于290000,分离度大于2.8;五味子醇甲的理论塔板数大于140000,分离度大于1.5;故规定理论塔板数以芍药苷峰计不得低于15000。
表245种成分理论塔板数和分离度统计
7样品测定
取上述10批痰喘半夏颗粒样品,每批平行2份,按供试品制备方法制备,精密吸取20μl,注入液相色谱仪,按外标一点法计算各指标含量,结果见表25。10批样品中芍药苷、沉香四醇、甘草苷、甘草酸、五味子醇甲的平均含量分别为5.36、1.27、2.59、2.22、0.11mg/袋。
表25痰喘半夏颗粒中5种指标成分的样品测定结果
8含量限度制定
五味子醇甲的平均含量为0.037mg/g,即0.0037%,相当于37个ppm,由于含量过低,故不上正文,以薄层鉴别即可。其余4种成分按照10批样品所得含量测定结果的80%来规定限度:
本品每袋含白芍以芍药苷(c23h28o11)计,不得少于4.3mg;
每袋含沉香以沉香四醇(c17h18o6)计,不得少于1.0mg;
每袋含甘草以甘草酸(c42h62o16)计,不得少于1.8mg,以甘草苷(c21h22o9)计,不得少于2.0mg。
根据以上规定,10批样品全部符合本发明的规定。
虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作出一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!