高灵敏的Fe-PANI/Pt@Au多巴胺电化学检测电极及其制备的制作方法
本发明涉及一种高灵敏的fe-pani/pt@au多巴胺电化学检测电极及其制备。
背景技术:
多巴胺(da)是一种存在于中枢神经、心血管、肾脏及内分泌系统中的儿茶酚胺类神经递质,且在人体中会对人们的思考、工作、运动等行为产生影响。多巴胺在人体中的正常含量为0.2-0.4g/ml,其在人脑中特定区域的含量分布影响垂体内分泌机能的协调。当人体中多巴胺的含量过低时会引起如精神分裂症、帕金森症、心脏衰竭、神经肌肉失调等疾病。反之da含量过多,会让人感到兴奋、开心,容易上瘾。另外在医学方面,多巴胺也常被用于治疗肾功能衰竭、抑郁症、以及内毒素败血症等疾病。因此,开展多巴胺分析检测方法在研究神经生理学及相关药物的质量控制方面具有重要的意义,有选择性地、高灵敏度地检测神经递质多巴胺的浓度,对在分子水平上诊断这些疾病有重要的应用价值。
由于多巴胺检测的重要性科学家们一直在致力于多巴胺检测的研究来获得高灵敏的、选择性的、快速的多巴胺检测技术方法。目前多巴胺常用的检测方法有荧光光谱法、高校液相法、毛细管电泳法等,而电化学检测方法由于具有易操作、高灵敏、选择性好等优点,成为多巴胺检测的常用方法。然在大脑和体液中,多巴胺、抗坏血酸、尿酸的氧化还原峰电位比较相近限制了电化学检测da技术,采用修饰电极可以增加da的传质速率,提高测定的灵敏度与抗干扰能力,因此许多科研人员研究使用修饰电极对电化学方法来进行改良。
近年来,金属及高分子导电聚合物在纳米范围内形成的金属/导电聚合物复合纳米材料因其特殊的功能和效应引起了人们的广泛关注。研究表明,将au、pt、ag、cu等纳米金属粒子引进聚苯胺,可以提高聚苯胺的导电性能和电化学响应性能,同时还保留了金属粒子原本的物理和化学性能。聚苯胺/金属复合纳米材料在电化学检测人体生物传感领域的研究已成为当今的一大热点。制备用于多巴胺高灵敏的、选择性检测的的聚苯胺/金属复合材料修饰电极具有较好的应用前景。
技术实现要素:
本发明提供了一种高灵敏的fe-pani/pt@au多巴胺电化学检测电极及其制备,可以有效解决多巴胺检测过程中的灵敏度及尿酸、抗坏血酸的干扰问题。
本发明是这样实现的:
一种高灵敏的fe-pani/pt@au多巴胺电化学检测电极的制备方法,包括以下步骤:
s1,将0.5ml苯胺单体加入50ml二氯甲烷溶液中超声使其均匀分散形成透明均一的有机相溶液;
s2,在搅拌条件下往所述有机相溶液中依次缓慢加入0.103ml的24.28mm的氯金酸溶液、50ml的0.2mol/l的n-甲基吡咯烷酮水溶液与0.05ml的0.5mol/l的氯化铁溶液,室温下避光搅拌一段时间后静置;
s3,将0.3g过硫酸铵加入8.6ml的2mol/l的盐酸溶液中形成过硫酸铵-盐酸溶液,将过硫酸铵-盐酸溶液缓慢加入步骤s2中反应溶液中,静置反应一段时间后用有机系滤膜过滤得到固体产物;
s4,将所述固体产物清洗、过滤、自然晾干得到fe-pani/au粉体;
s5,取25~35mg的fe-pani/au粉体分散于15-18.2mω纯水中,在搅拌条件下依次加入0.9ml的8.228mm的氯铂酸溶液和0.3ml的100mm抗坏血酸溶液,在室温下避光搅拌反应10-30h;
s6,反应结束后,取0.5ml浓盐酸加入s5的反应溶液中搅拌一段时间,然后用0.22μm孔径的水系滤膜过滤得到固体产物,将所述固体产物清洗、过滤、自然晾干得到fe-pani/pt@au粉体;
s7,将所述玻碳电极依次用3μm、1μm、0.3μm、50nm的氧化铝微粉研磨抛光后备用;
s8,取1~3mg的fe-pani/pt@au粉体超声分散于0.5ml无水乙醇中,加入5~15μl的nafion溶液超声分散使其形成分散均匀的悬浮液;
s9,取10-40μl的悬浮液以10-20μl/次滴加到步骤s7抛光后的玻碳电极表面,静置待溶液挥发后继续滴加悬浮液,反复操作最终得到fe-pani/pt@au修饰电极。
本发明还进一步提供一种通过上述方法获得的高灵敏的fe-pani/pt@au多巴胺电化学检测电极;其测试环境为:以fe-pani/pt@au修饰电极为工作电极、石墨电极或铂电极为对电极、饱和甘共电极为参比电极进行dpv测试,其中,电位范围-0.1v-0.9v(vs.sce),扫描振幅为50mv,脉冲宽度50ms,整个测试环境在ph=6.6-7.2的磷酸缓冲液中进行。
本发明的有益效果是:fe-pani/pt@au纳米复合材料修饰电极用于da的检测时,在ua存在的条件下,不会对da的检测造成干扰;同时aa在修饰电极上电化学响应较弱,对检测体系没有造成干扰。体系对da检出限为0.001μm。对ua为检测限为0.1μm。。
附图说明
为了更清楚地说明本发明实施方式的技术方案,下面将对实施方式中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
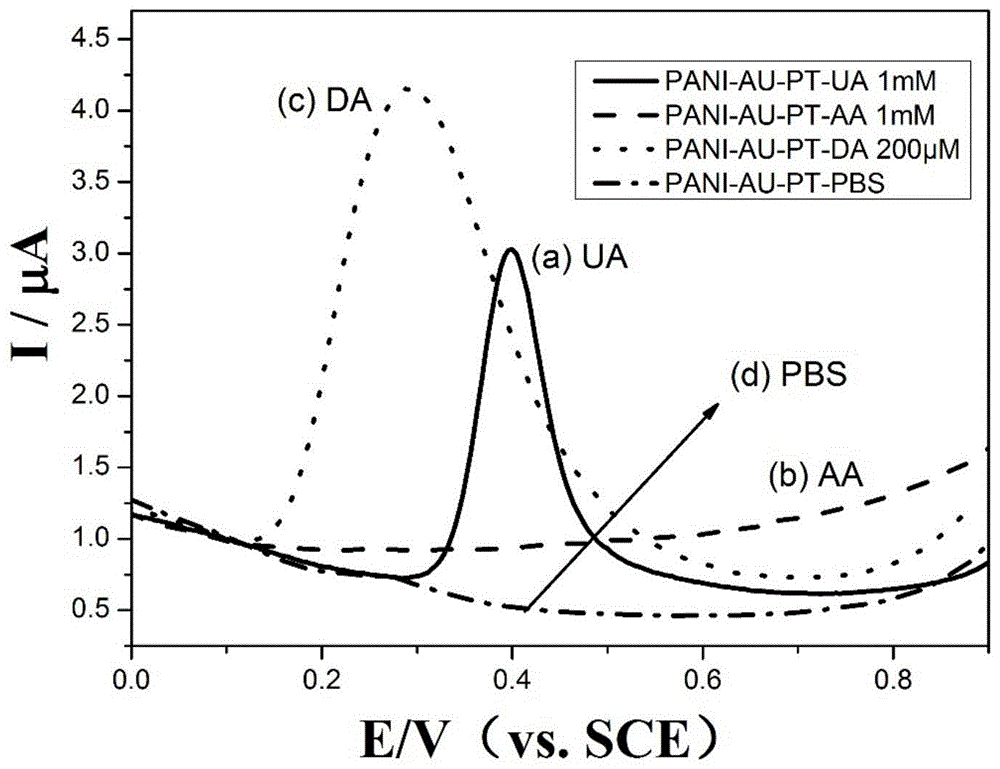
图1是在0.1m,ph为6.8的磷酸缓冲液中,1mm的ua、2mm的aa、200μm的da在pani/pt@au修饰电极上的dpv图。图2是在0.1m,ph为6.8的磷酸缓冲液中,1mm的ua、1mm的aa、200μm的da在fe-pani/pt修饰电极上的dpv图。
图3是在浓度为0.1m,ph为6.8的磷酸缓冲液中1mm的ua、2mm的aa、200μm的da在fe-pani/pt@au修饰电极上的dpv图。
图4是在浓度为0.1m,ph为6.8的磷酸缓冲液中,不同浓度da在fe-pani/pt@au修饰电极上的dpv图。
图5为da浓度与峰电流的线性关系曲线图。
图6是在浓度为0.1m,ph为6.8的磷酸缓冲液中,一系列浓度ua在fe-pani/pt@au修饰电极上的dpv图。
图7为ua浓度与峰电流的线性关系曲线图。
具体实施方式
为使本发明实施方式的目的、技术方案和优点更加清楚,下面将结合本发明实施方式中的附图,对本发明实施方式中的技术方案进行清楚、完整地描述,显然,所描述的实施方式是本发明一部分实施方式,而不是全部的实施方式。基于本发明中的实施方式,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施方式,都属于本发明保护的范围。因此,以下对在附图中提供的本发明的实施方式的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施方式。基于本发明中的实施方式,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施方式,都属于本发明保护的范围。
本发明实施例提供一种高灵敏的fe-pani/pt@au多巴胺电化学检测电极的制备方法,包括以下步骤:
s1,将0.5ml苯胺单体加入50ml二氯甲烷溶液中超声使其均匀分散形成透明均一的有机相溶液;
s2,在搅拌条件下往所述有机相溶液中依次缓慢加入0.103ml的24.28mm的氯金酸溶液、50ml的0.2mol/l的n-甲基吡咯烷酮水溶液与0.05ml的0.5mol/l的氯化铁溶液,室温下避光搅拌一段时间后静置;
s3,将0.3g过硫酸铵加入8.6ml的2mol/l的盐酸溶液中形成过硫酸铵-盐酸溶液,将过硫酸铵-盐酸溶液缓慢加入步骤s2中反应溶液中,静置反应一段时间后用有机系滤膜过滤得到固体产物;
s4,将所述固体产物清洗、过滤、自然晾干得到fe-pani/au粉体;
s5,取25~35mg的fe-pani/au粉体分散于15-18.2mω纯水中,在搅拌条件下依次加入0.9ml的8.228mm的氯铂酸溶液和0.3ml的100mm抗坏血酸溶液,在室温下避光搅拌反应10-30h;
s6,反应结束后,取0.5ml浓盐酸加入s5的反应溶液中搅拌一段时间,然后用0.22μm孔径的水系滤膜过滤得到固体产物,将所述固体产物清洗、过滤、自然晾干得到fe-pani/pt@au粉体;
s7,将所述玻碳电极依次用3μm、1μm、0.3μm、50nm的氧化铝微粉研磨抛光后备用;
s8,取1~3mg的fe-pani/pt@au粉体超声分散于0.5ml无水乙醇中,加入5~15μl的nafion溶液超声分散使其形成均匀的悬浮液;
s9,取10-40μl的悬浮液以10-20μl/次滴加到步骤s7抛光后的玻碳电极表面,静置待溶液挥发后继续滴加悬浮液,反复操作最终得到fe-pani/pt@au修饰电极。
所述fe-pani/pt@au修饰电极的测试环境为:以fe-pani/pt@au修饰电极为工作电极、石墨电极或铂电极为对电极、饱和甘共电极为参比电极进行dpv测试,其中,电位范围-0.1v-0.9v(vs.sce),扫描振幅为50mv,脉冲宽度50ms,整个测试环境在ph=6.6-7.2发磷酸缓冲液中进行。作为进一步改进的,在步骤s2中,混合溶液避光反应时间为10-50h。作为进一步改进的,另外,在整个测试中,环境的ph值对测试也具有较大影响,当ph=6.6-7.2超过这一范围时,聚苯胺的质子掺杂程度发生变化,从而影响检测物质的抗干扰性、灵敏度及稳定性,因此优选的,整个测试环境在ph=6.8-7.2的磷酸缓冲液中进行。
作为进一步改进的,在步骤s3中,加入过硫酸铵-盐酸溶液后静置反应时间为15-35min。
作为进一步改进的,在步骤s4中,将固体产物清洗的步骤包括:
s41,将所述固体产物依次用蒸馏水、盐酸溶液、乙醇洗涤清洗过滤产物,盐酸溶液的浓度范围为0.1-2mol/l。在步骤中s41中,盐酸溶液清洗使得聚苯胺保持一定的导电性和分散性。然过高溶度的盐酸清洗溶液将使得聚苯胺易分散成均匀溶液,影响产品的清洗并破坏过滤膜层。故,更优选的,所述盐酸溶液的浓度范围为0.1-1.2mol/l。
作为进一步改进的,在步骤s5中,取25~35mg的fe-pani/au粉体分散于15-18.2mω纯水中,在搅拌条件下依次加入0.9ml的8.228mm的氯铂酸溶液和0.3ml的100mm抗坏血酸溶液,在室温下避光搅拌反应10-30h的步骤包括:
s51,取30mg的fe-pani/au粉体分散于18.2mω纯水中,在搅拌条件下依次加入0.9ml的8.228mm的氯铂酸溶液和0.3ml的100mm抗坏血酸溶液,在室温下避光搅拌反应20h。
作为进一步改进的,在步骤s6中,反应结束后,取0.5ml浓盐酸加入s5的反应溶液中搅拌反应时间为20min-1h。
作为进一步改进的,在步骤s6中,将固体产物清洗的步骤包括:
s61,将所述固体产物依次用蒸馏水、盐酸溶液、乙醇洗涤清洗过滤产物,盐酸溶液的浓度范围为0.1-2mol/l。
作为进一步改进的,在步骤s8中,所述取1~3mg的fe-pani/pt@au粉体超声分散于0.5ml无水乙醇中,加入5~15μl的nafion溶液超声分散使其形成均匀的悬浮液的步骤包括:
s81,取2mg的fe-pani/pt@au粉体超声分散于0.5ml无水乙醇中,加入10μl的nafion溶液超声分散使其形成均匀的悬浮液。
作为进一步改进的,在步骤s9中,所述取10-40μl的悬浮液以10-20μl/次滴加到步骤s7抛光后的玻碳电极表面,静置待溶液挥发后继续滴加悬浮液,反复操作最终得到fe-pani/pt@au修饰电极的步骤包括:
s91,取20μl的悬浮液直接滴加到步骤s5抛光后的玻碳电极表面,静置待溶液挥发后得到fe-pani/pt@au修饰电极。
实施例:
将50ml二氯甲烷加入到圆底烧瓶中,再将0.5ml苯胺单体加入二氯甲烷溶液中超声使其均匀分散形成透明均一的有机相溶液。将得到的有机相溶液置放与磁力搅拌器上,在搅拌条件下往溶液中缓慢加入0.103ml24.28mm的氯金酸溶液、50ml0.2mol/l的n-甲基吡咯烷酮水溶液与0.05ml0.5mol/l的氯化铁的混合溶液,室温下避光搅拌20h后静置。称取0.3g过硫酸铵溶于8.6ml2mol/l的盐酸溶液中,完全溶解后得到过硫酸铵-盐酸溶液。将过硫酸铵-盐酸混合液沿着圆底烧瓶瓶壁缓慢加入静置条件下的二氯甲烷-n-甲基吡咯烷酮反应溶液中,静置反应20分钟后用0.22μm孔径的有机系滤膜过滤得到固体产物,依次用蒸馏水、1mol/l盐酸溶液、乙醇洗涤清洗过滤产物,自然晾干得到fe-pani/au粉体。
取fe-pani/au粉体30mg,用75ml18.2mω纯水分散于150ml的圆底烧瓶中,在搅拌条件下依次加入0.9ml8.228mm的氯铂酸溶液和0.3ml100mm抗坏血酸溶液,在室温下避光搅拌反应20h。反应结束后,取0.5ml浓盐酸加入反应溶液中,搅拌20min后用0.22μm孔径的水系滤膜过滤得到固体产物,依次用蒸馏水、0.1m盐酸、乙醇洗涤,自然晾干得到fe-pani/pt@au粉体。将直径为5mm的玻碳电极依次用3μm、1μm、0.3μm、50nm的氧化铝微粉研磨抛光后备用。取2mgfe-pani/pt@au粉体超声分散于0.5ml无水乙醇中,加入10μl的nafion溶液超声分散30min使其形成均匀的悬浮液。取20μl的悬浮液滴加到干净的玻碳电极表面,静置待溶液挥发后得到fe-pani/pt@au修饰电极待用。以fe-pani/pt@au修饰电极为工作电极、石墨电极为对电极、饱和甘共电极为参比电极进行脉冲伏安(dpv)测试。其中电位范围-0.1v-0.9v(vs.sce),扫描振幅为50mv,脉冲宽度50ms。整个实验均在磷酸缓冲液(0.1mpbs溶液,ph=6.8)的环境中进行。
对比例1:
将50ml二氯甲烷加入到圆底烧瓶中,再将0.5ml苯胺单体加入二氯甲烷溶液中超声使其均匀分散形成透明均一的有机相溶液。将得到的有机相溶液置放与磁力搅拌器上,在搅拌条件下往溶液中缓慢加入0.103ml24.28mm的氯金酸溶液与50ml0.2mol/l的n-甲基吡咯烷酮水溶液的混合溶液,室温下避光搅拌20h后静置。称取0.3g过硫酸铵溶于8.6ml2mol/l的盐酸溶液中,完全溶解后得到过硫酸铵-盐酸溶液。将过硫酸铵-盐酸混合液沿着圆底烧瓶瓶壁缓慢加入静置条件下的二氯甲烷-n-甲基吡咯烷酮反应溶液中,静置反应20分钟后用0.22μm孔径的有机系滤膜过滤得到固体产物,依次用蒸馏水、1mol/l盐酸溶液、乙醇洗涤清洗过滤产物,自然晾干得到pani/au粉体。
取pani/au粉体30mg,用75ml18.2mω纯水分散于150ml的圆底烧瓶中,在搅拌条件下依次加入0.9ml8.228mm的氯铂酸溶液和0.3ml100mm抗坏血酸溶液,在室温下避光搅拌反应20h。反应结束后,取0.5ml浓盐酸加入反应溶液中,搅拌20min后用0.22μm孔径的水系滤膜过滤得到固体产物,依次用蒸馏水、0.1m盐酸、乙醇洗涤,自然晾干得到pani/pt@au粉体。将直径为5mm的玻碳电极依次用3μm、1μm、0.3μm、50nm的氧化铝微粉研磨抛光后备用。取2mgpani/pt@au粉体超声分散于0.5ml无水乙醇中,加入10μl的nafion溶液超声分散30min使其形成均匀的悬浮液。取20μl的悬浮液滴加到干净的玻碳电极表面,静置待溶液挥发后得到pani/pt@au修饰电极待用。以pani/pt@au修饰电极为工作电极、石墨电极为对电极、饱和甘共电极为参比电极进行脉冲伏安(dpv)测试。其中电位范围-0.1v-0.9v(vs.sce),扫描振幅为50mv,脉冲宽度50ms。整个实验均在磷酸缓冲液(0.1mpbs溶液,ph=6.8)的环境中进行。
对比例2:
将50ml二氯甲烷加入到圆底烧瓶中,再将0.5ml苯胺单体加入二氯甲烷溶液中超声使其均匀分散形成透明均一的有机相溶液。将得到的有机相溶液置放与磁力搅拌器上,在搅拌条件下往溶液中依次缓慢加入50ml0.2mol/l的n-甲基吡咯烷酮水溶液与0.05ml0.5mol/l的氯化铁溶液,室温下避光搅拌20h后静置。称取0.3g过硫酸铵溶于8.6ml2mol/l的盐酸溶液中,完全溶解后得到过硫酸铵-盐酸溶液。将过硫酸铵-盐酸混合液沿着圆底烧瓶瓶壁缓慢加入静置条件下的二氯甲烷-n-甲基吡咯烷酮反应溶液中,静置反应20分钟后用0.22μm孔径的有机系滤膜过滤得到固体产物,依次用蒸馏水、1mol/l盐酸溶液、乙醇洗涤清洗过滤产物,自然晾干得到fe-pani粉体。取铁掺杂聚苯胺fe-pani纳米材料30mg,用75ml18.2mω纯水分散于150ml的圆底烧瓶中,在搅拌条件下依次加入0.9ml8.228mm的氯铂酸溶液和0.3ml100mm抗坏血酸溶液,在室温下避光搅拌反应20h。反应结束后,取0.5ml浓盐酸加入反应溶液中,搅拌20min后用0.22μm孔径的水系滤膜过滤得到固体产物,依次用蒸馏水、0.1m盐酸、乙醇洗涤,自然晾干得到铁掺杂聚苯胺/铂(fe-pani/pt)粉体。将直径为5mm的玻碳电极依次用3μm、1μm、0.3μm、50nm的氧化铝微粉研磨抛光后备用。取2mgfe-pani/pt粉体超声分散于0.5ml无水乙醇中,加入10μl的nafion溶液超声分散30min使其形成均匀的悬浮液。取20μl的悬浮液滴加到干净的玻碳电极表面,静置待溶液挥发后得到fe-pani/pt修饰电极待用。以fe-pani/pt修饰电极为工作电极、石墨电极为对电极、饱和甘共电极为参比电极进行脉冲伏安(dpv)测试。其中电位范围-0.1v-0.9v(vs.sce),扫描振幅为50mv,脉冲宽度50ms。整个实验均在磷酸缓冲液(0.1mpbs溶液,ph=6.8)的环境中进行。
试验例:
图1是在0.1m,ph为6.8的磷酸缓冲液中,1mm的ua、2mm的aa、200μm的da在pani/pt@au修饰电极上的dpv图,从图可以看出,da的氧化电位位于0.15v-0.6v,氧化峰0.29v;ua的氧化电位位于0.3v-0.6v,氧化峰0.4v;da与ua氧化信号存在较大程度的交叠,因此ua的存在会对da的检测造成干扰。在体系的测试条件下,aa没有表现出明显的电化学响应信号,对体系da或ua的检测没有干扰。另,pani/pt@au修饰电极对da的检出限为1μm。
图2是在0.1m,ph为6.8的磷酸缓冲液中,1mm的ua、1mm的aa、200μm的da在fe-pani/pt修饰电极上的dpv图,从图可以看出,da的氧化电位位于0.2v-0.6v之间,峰电位为0.425v,而ua和aa在电位范围区间均没有表现出明显的氧化信号。另,其对da的检出限为1μm。
图3是在浓度为0.1m,ph为6.8的磷酸缓冲液中1mm的ua、2mm的aa、200μm的da在fe-pani/pt@au修饰电极上的dpv图,由图可以看出在fe-pani-au-pt修饰电极上da的氧化电位位于0.15-0.3v之间,峰电位为0.23v,ua的氧化电位位于0.3-0.45v,峰电位为0.36v。由于da和ua在电极上的dpv氧化信号没有出现明显的交叠,因此体系在ua存在的条件下,不会对da的检测造成干扰;同时aa在修饰电极上电化学响应较弱,对检测体系没有造成干扰。
图4是在浓度为0.1m,ph为6.8的磷酸缓冲液中,不同浓度da在fe-pani/pt@au修饰电极上的dpv图,可以看出da的氧化电位位于0.15v-0.35v之间,峰电位为0.23v。随着da浓度的增大,电极上的响应电流也随着增大,检出限为0.001μm。
图5为da浓度与峰电流的线性关系曲线图。从图中可以看出,在文中检出的浓度范围0.001μm-200μm,da浓度与峰电流之间呈现双线性关系。在高浓度(10μm-200μm)线性曲线为y1=0.246x+3.64034,其相关系数r2=0.99785。在低浓度(0.001μm-10μm)线性曲线为y2=0.58923x+0.41035,其相关系数r2=0.96184。
图6是在浓度为0.1m,ph为6.8的磷酸缓冲液中,一系列浓度ua在fe-pani/pt@au修饰电极上的dpv图,图中ua氧化电位位于0.25v-0.45v之间,峰位电位为0.36v。随着ua浓度的增大,电极上的响应电流也随着增大,检测限为0.1μm。
图7为ua浓度与峰电流的线性关系曲线图。从图中可以看出,在文中检出的浓度范围0.1μm-1mm,ua浓度与峰电流之间呈现双线性关系。在高浓度(10μm-1mm)线性曲线为y1=0.03187x+3.32917,其相关系数r2=0.99843。在低浓度(0.1μm-10μm)线性曲线为y2=0.24969x+0.51304,其相关系数r2=0.97423。
以上所述仅为本发明的优选实施方式而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!