分离纯化达巴万星前体A40926的方法与流程
本发明涉及生物
技术领域:
,具体地,本发明涉及分离纯化达巴万星前体a40926的方法。
背景技术:
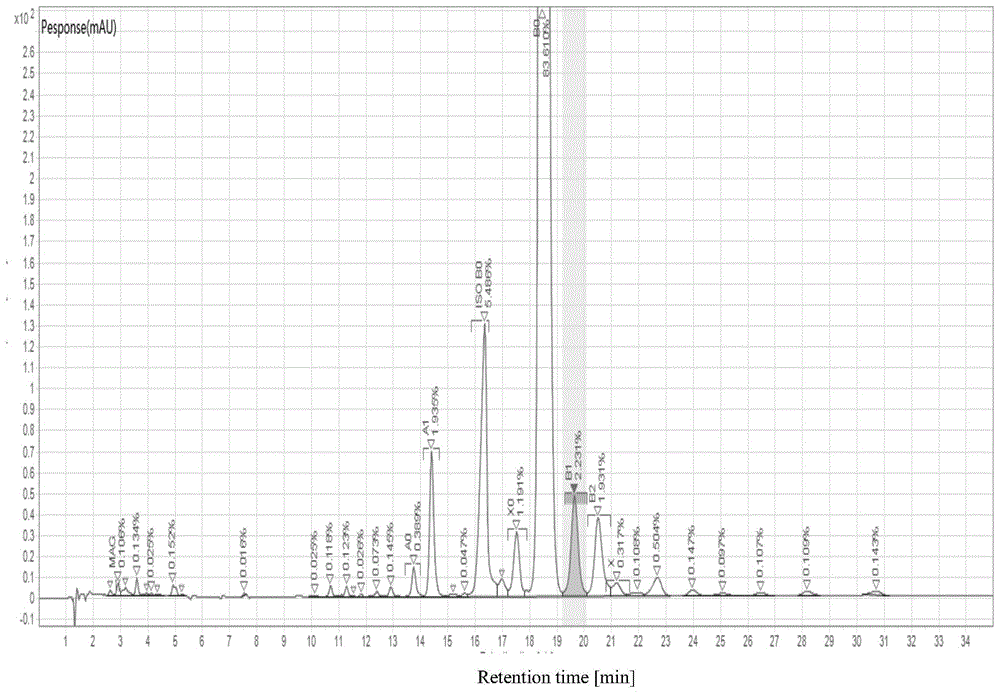
:达巴万星(dalbavancin),也称作道古霉素,由天然糖肽类抗生素a40926(结构和活性与替考拉宁类似)经过对末端羧基的酰胺化修饰衍生而成的一类新型半合成糖肽类抗生素。而a40926-b0是达巴万星合成的主要前体物质。a40926是由野村放线菌属atcc39727菌株发酵产生,a40926是多组分化合物,含有5个主要组分:pa、pb、a、b0和b1,b0和b1统称为b组分,其中b0占90%。这5组分均含有2个氯原子和2个糖基——甘露糖和葡糖胺。结构与替考拉宁相似,在葡糖胺上有一脂肪酸链,pa和a中是正十一烷基,pb和b中是异十二烷基。结构中有2个羧酸,因此在中性条件下带有净负电荷。a、b和pa、pb的区别在于甘露糖c-6位上的乙酰基。a40926个主要组分的结构如下所示:发酵液中pa、pb含量占优,经下游提取工艺碱性环境中水解后,pa、pb会慢慢脱乙酰化为a、b组分。因此,a、b组分为主要活性研究组分。技术实现要素:本申请是基于发明人对以下事实和问题的发现和认识作出的:发明人发现,现有技术中,a40926生物发酵生产多组分化合物,除了主要组分pa、pb、a、b0和b1,还含有众多组分异构杂质或者类似物杂质,比如,b0组分异构体isob0,降解杂质mag,尤其与主峰b0靠近的杂质很难用一般层析法纯化去除,同时,现有技术中,对单杂水平未有报道。本领域技术人员所了解的是,对于产品单杂控制线在0.15%以下仅通过定性手段(例如液相色谱相对保留时间)来定义杂质,无需确定其结构,而高于界定限的杂质需要界定其安全性。因此,降低杂质至鉴定限以下,这对于产品工艺开发以及杂质研究都具有重要意义。在本发明的第一方面,本发明提出了一种分离纯化a40926的方法。根据本发明的实施例,所述方法包括以下步骤:通过高压层析-hplc高效液相色谱对a40926样品进行分离,收集b0峰值的洗脱液,以便获得b0洗脱液。发明人发现,通过对a40926样品进行高压层析-hplc高效液相色谱分离,所获得的b0洗脱液中b0纯度可达97%,任意单杂含量<0.15%。根据本发明的实施例,上述方法还可以进一步包括如下附加技术特征至少之一:根据本发明的实施例,所述高压层析-hplc高效液相色谱采用下列条件:色谱柱填料粒径为5-20μm,50-300a孔径的高分子微球;色谱柱填料吸附载量3-20g/l;流动相包含缓冲盐和有机溶剂,所述缓冲盐的浓度为25~35mmol/l,优选,30mmol/l,所述流动相的ph为5~10,优选8;流速为每分钟1~180ml,优选1~90ml;检测波长为280nm。等度洗脱。发明人发现,色谱柱填料选择粒径更细、分辨率更高的高分子微球层析介质,可大大提升单杂的分离效果,提高提取物中b0纯度和降低单杂的含量;同时,发明人发现,流动相中缓冲盐的浓度、流动相的ph影响提取物中b0纯度和单杂的含量。采用上述条件对a40926进行高压层析液相色谱分离,可进一步提高b0纯度以及降低单杂含量。根据本发明的实施例,所述有机溶剂为甲醇、丙酮、异丙醇、乙醇或乙腈,优选,乙腈。根据本发明的实施例,所述缓冲盐为乙酸铵、tris或磷酸盐,优选,磷酸盐,更优选,磷酸二氢钠。根据本发明的实施例,所述等度洗脱是通过如下方式进行的:采用包含70~75%流动相a和25~30%流动相b的流动相冲洗6~10个柱体积。根据本发明的具体实施例,所述等度洗脱是通过如下方式进行的:采用包含70~75%30mmol/l的磷酸二氢钠和25~30%乙腈的流动相冲洗6~10个柱体积,优选地,所述乙腈的浓度为26%。在上述等度洗脱的方式下,6-10个柱体积就可完成达巴万星前体a40926主峰的洗脱,本过程所用流动相量较少,纯化周期比较短。根据本发明的实施例,上样前,采用包含10~20%去离子水和80~90%流动相b的流动相冲洗5~10个柱体积,以再生色谱柱;采用包含90~95%流动相a和5~10%流动相b的流动相冲洗5~10个柱体积,以平衡色谱柱。根据本发明的具体实施例,上样前,采用包含10~20%去离子水和80~90%乙腈的流动相冲洗5~10个柱体积,以再生色谱柱;采用包含90~95%30mmol/l的磷酸二氢钠和和5~10%乙腈的流动相冲洗5~10个柱体积,以平衡色谱柱。上样前,对层析柱进行柱前预处理,先用高浓度有机相进行再生活化色谱填料,后用浓度低于10%的有机相进行平衡,可避免色谱柱未再生干净造成洗脱杂质出峰异常。根据本发明的实施例,所述色谱柱为c18色谱柱。具体地,所述色谱柱为赛芬c18,polyrp-300,lunac18,welchxtimtec18,watersxbridgec18或纳微unisil10-300c18;优选地,所述色谱柱为赛芬c18或lunac18。根据本发明的实施例,所述a40926样品预先溶解于浓度为5%-10%(v/v)的乙腈溶液中,所述溶液ph为4~8。根据本发明的实施例,采用分段收集的方式收集b0峰值的洗脱液,以便获得不同段洗脱液;合并b0含量不低于97%,任意单杂含量<0.15%的段洗脱液,以便获得所述b0洗脱液。进而可进一步提高所获得的b0洗脱液中b0的纯度和降低任意单杂的含量。在本发明的第二方面,本发明提出了一种分离纯化a40926的方法。根据本发明的实施例,所述方法包括:称取a40926粗品,配置30mg/ml的样品,其中b080~85%,isob05~6%,单杂≤2%,加入10%乙腈,样品完全溶解后,用氢氧化钠调节样品ph5.0,超声脱气处理,用孔径为0.45μm滤膜处理样品,收集滤液待用;选用赛芬c18高分子微球层析填料,粒径10μm,孔径100a,装柱4.6*250mm,柱体积4ml;用85%乙腈水溶液1ml/min的流速再生c18填料10个柱体积,上样前先用ph5.0乙腈浓度10%流动相1ml/min的流速平衡色谱柱5cv,然后用1ml/min的流速上样,上样载量15g/l;上样后用ph5.0乙腈浓度10%的流动相冲洗色谱柱;采用ph8.030mmol/l的磷酸二氢钠水溶液和乙腈有机溶剂作为流动相进行等度洗脱,流速控制在1ml/min,洗脱直接用乙腈浓度26%的流动相冲洗10个柱体积洗脱a40926主峰至峰下降,再用乙腈含量为85%的水溶液冲洗3个柱体积洗脱后杂;b0出峰开始收样,分段收集洗脱液,洗脱至峰下降结束收样,对符合b0>97%,任意单杂<0.15%要求的段洗脱液进行合并取样。根据本发明实施例的上述分离纯化a40926的方法,针对现有纯化工艺存在的不足,提供一种高效分离纯化去除杂质的方法,仅需一步高压层析即可纯化分离任意单杂<0.15%,纯化a40926主要组分b0含量高于97%。本发明分离方法简单,可规模化生产,高压层析载量可达15g/l,层析纯化收率80%以上,高载量高收率。在本发明的第三方面,本发明提出了一种a40926。根据本发明的实施例,b0含量不低于97%,任意单杂含量<0.15%。在本发明的第四方面,本发明提出了一种a40926。根据本发明的实施例,所述a40926是经过前面所述的方法分离纯化获得的。附图说明图1是根据本发明实施例的a40926粗品的高效液相色谱分析图;图2是根据本发明实施例1的c18高压层析洗脱液样品的高效液相色谱分析图;图3是根据本发明实施例2的c18高压层析洗脱液样品的高效液相色谱分析图;图4是根据本发明实施例3的c18高压层析洗脱液样品的高效液相色谱分析图;图5是根据本发明实施例12的聚合物层析洗脱液样品的的高效液相色谱分析图。具体实施方式下面详细描述本发明的实施例,所述实施例的示例在附图中示出。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。实施例1称取a40926粗品(粗品的高效液相色谱分析如图1所示),配置30mg/ml(以b0浓度计)的样品,其中b083.61%,isob05.48%,单杂≤1.91%。加入10%乙腈有机相,样品完全溶解后,用氢氧化钠调节样品ph5.0,超声脱气处理。用孔径为0.45μm滤膜处理样品,收集滤液待用。选用赛芬c18(赛芬科技有限公司生产)高分子微球层析填料,粒径10μm,孔径100a,装柱4.6*250mm,柱体积4ml。用85%乙腈水溶液1ml/min的流速再生c18填料10cv。上样前先用ph5.0乙腈浓度10%流动相1ml/min的流速平衡色谱柱5cv,然后用1ml/min的流速上样,上样载量15g/l(按b0浓度计)。上样后用ph5.0乙腈浓度10%的流动相冲洗色谱柱。采用ph8.030mmol/l的磷酸二氢钠水溶液和乙腈有机溶剂作为流动相进行等度洗脱,流速控制在1ml/min。洗脱直接用乙腈浓度26%的流动相冲洗10个柱体积洗脱a40926主峰至峰下降,再用乙腈含量为85%的水溶液冲洗3个柱体积洗脱后杂。分段收集目的峰值的溶液,对符合要求的组份液进行合并取样,经hplc高效液相色谱分析,洗脱液中达巴万星b0的纯度97.80%,单杂0.03%,如附图2,层析收率85%。实施例2称取a40926粗品,配置50mg/ml(以b0计)的样品,加入5%乙腈有机相,样品完全溶结后,用氢氧化钠调节样品ph5.0,超声脱气处理。用孔径为0.45μm滤膜处理样品,收集滤液待用。选用unisil10-300c18(纳微科技有限公司生产)高分子微球层析填料,装柱dac50×20mm柱500ml。用85%乙腈溶液100ml/min再生c18填料10bv,然后用ph5.0低浓度的乙腈溶液冲洗色谱柱。上样前先用浓度10%的乙腈相100ml/min的流速平衡色谱柱5bv,然后用80ml/min的流速上样,上样载量7.8g/l。上样后用ph5.0浓度10%的乙腈相进行冲洗色谱柱。采用ph8.030nm的乙酸铵水溶液和乙腈溶液作为流动相进行等度洗脱,流速控制在90ml/min。洗脱直接用30%的流动相冲洗10个柱体积洗脱主峰,再用乙腈含量为85%的流动相冲洗3个柱体积洗脱后杂。分段收集目的峰值的溶液,对符合要求的组份液进行合并取样,经hplc高效液相色谱分析,洗脱液中达巴万星b0的纯度97%,单杂0.08%,如附图3,层析收率80%。实施例3称取a40926粗品,配置30mg/ml(以b0计)的样品,加入5%乙腈有机相,样品完全溶结后,用氢氧化钠调节样品ph6.0,超声脱气处理。待溶液澄清后,用孔径为0.45μm滤膜处理样品,收集滤液待用。选用赛芬c18(赛芬科技有限公司生产)高分子微球层析填料,装柱dac50×20mm柱500ml。用85%乙腈溶液100ml/min再生c18填料10bv,然后用低浓度的乙腈溶液冲洗色谱柱。上样前先用ph6.0浓度10%的乙腈相100ml/min的流速平衡色谱柱5bv,然后用80ml/min的流速上样,上样载量3.5g/l。上样后用ph6.0浓度10%的乙腈相进行冲洗色谱柱。采用30nm的磷酸二氢钠水溶液和乙腈有机溶剂作为流动相进行等度洗脱,流速控制在100ml/min。洗脱直接用ph8.030%的流动相冲洗10个柱体积洗脱主峰,再用乙腈含量为85%的流动相冲洗3个柱体积洗脱后杂、再生填料。分段收集目的峰值的溶液,对符合要求的组份液进行合并取样,经hplc高效液相色谱分析,洗脱液中达巴万星b0的纯度95%,单杂0.027%,如附图4,层析收率85%。实施例4称取a40926粗品,配置10mg/ml(以b0计)的样品,加入5%乙醇有机相,样品完全溶结后,用氢氧化钠调节样品ph5.0,超声脱气处理。用孔径为0.45μm滤膜处理样品,收集滤液待用。选用unisil10-300c18(纳微科技有限公司生产)高分子微球层析填料,装柱4.6*250mm柱4ml。用95%乙醇溶液1ml/min再生c18填料10bv,然后用浓度<50%的乙醇溶液冲洗色谱柱。上样前先用5%乙醇浓度流动相1ml/min的流速平衡色谱柱5bv,然后用1ml/min的流速上样,上样载量15g/l(按b0浓度计)。上样后用ph5.0浓度5%乙醇浓度的流动相冲洗色谱柱。采用ph6.530mm的磷酸二氢钠水溶液和乙腈溶液作为流动相进行等度洗脱,流速控制在1ml/min。洗脱直接用27%的流动相冲洗10个柱体积洗脱主峰,再用乙醇含量为95%的流动相冲洗3个柱体积洗脱后杂。分段收集目的峰值的溶液,对符合要求的组份液进行合并取样,经hplc高效液相色谱分析,洗脱液中达巴万星b0的纯度97%,单杂0.10%,层析收率80%。实施例5工艺放大称取a40926粗品,配置50mg/ml(以b0计)的样品,其中b083.61%,isob05.48%,单杂≤1.91%。加入5%乙腈有机相,样品完全溶解后,用氢氧化钠调节样品ph5.0,超声脱气处理。用孔径为0.45μm滤膜处理样品,收集滤液待用。选用luna10umprepc18(菲罗门科技有限公司生产,n0:00g-4616-e0)高分子微球层析填料,粒径10um,孔径100a,装柱dac50×25cm,柱体积500ml。用85%乙腈溶液100ml/min的流速再生c18填料10cv,然后用ph5.0低浓度的乙腈溶液冲洗色谱柱。上样前先用ph5.0乙腈浓度10%的流动相以100ml/min的流速平衡色谱柱5cv,然后用80ml/min的流速上样,上样载量10g/l。上样后用ph5.0乙腈浓度10%的流动相冲洗柱床。采用ph8.030mmol/l磷酸二氢钠水溶液和乙腈有机试剂作为流动相进行等度洗脱,流速控制在90ml/min。洗脱直接用乙腈浓度26%的流动相冲洗10个柱体积洗脱主峰至峰下降,再用乙腈含量为85%的流动相冲洗3个柱体积洗脱后杂。分段收集目的峰值的溶液,对符合要求的组份液进行合并取样,经hplc高效液相色谱分析,洗脱液中达巴万星前体b0的纯度97.94%,单杂0.08%,层析收率84%。实施例6方法步骤同实施例5,所不同的是上样载量15g/l。分段收集目的峰值的溶液,对符合要求的组份液进行合并取样,经hplc高效液相分析,洗脱液中达巴万星前体b0的纯度96.24,单杂0.10%,层析收率82%。实施例7方法步骤同实施例5,所不同的是上样载量20g/l。分段收集目的峰值的溶液,对符合要求的组份液进行合并取样,经hplc高效液相分析,洗脱液中达巴万星前体b0的纯度94.8,单杂0.22%,层析收率75%。其中,实施例1、5~7的上样载量、柱体积、达巴万星前体b0的纯度、单杂含量、层析收率以及洗脱柱体积(cv)如下表1所示。表1:分析表1结果,对比实施例1载量15g/l纯化结果,dac50柱500ml放大载量20g/l时,纯化结果单杂>0.2%,b0纯度94.80%。dac50柱500ml载量在10g/l和15g/l时,与实施例1结果差异不大,b0纯度96%以上,单杂<0.2%,收率85%左右。实施例8方法步骤同实施例1,所不同的是洗脱直接用乙腈浓度28%的流动相冲洗10个柱体积洗脱a40926主峰至峰下降。洗脱液中达巴万星前体b0的纯度97%,单杂0.19%,层析收率80%实施例9方法步骤同实施例1,所不同的是洗脱直接用乙腈浓度30%的流动相冲洗10个柱体积洗脱a40926主峰至峰下降。洗脱液中达巴万星前体b0的纯度95.62%,单杂0.48%,层析收率81%。其中,实施例1、8~9的洗脱乙腈浓度、达巴万星前体b0的纯度、单杂含量、层析收率以及洗脱体积(cv)如下表2所示。表2:洗脱乙腈浓度%b0%单杂%收率%实施例12697.800.0385实施例82897.000.1980实施例93095.620.4881由表2可以看出,实施例1洗脱条件纯化a40926,b0纯度97.80%,单杂0.03%,层析收率85%。实施例8提高乙腈浓度至28%,b0纯度略有下降,单杂升高,洗脱收率降低。实施例9乙腈浓度进一步提高至30%时,b0纯度降低至95.62%,单杂0.48%超标。因此,洗脱流动相乙腈浓度影响洗脱后单纯的含量,最优为26%。实施例10方法步骤同实施例1,所不同的是采用ph7.030mmol/l的磷酸二氢钠水溶液和乙腈有机溶剂作为流动相进行等度洗脱。洗脱液中达巴万星前体b0的纯度97.09,单杂0.21,层析收率79%。实施例11方法步骤同实施例1,所不同的是采用ph9.030mmol/l的磷酸二氢钠水溶液和乙腈有机溶剂作为流动相进行等度洗脱。洗脱液中达巴万星前体b0的纯度95.30,单杂0.51,层析收率83%。其中,实施例1、10~11的洗脱ph、达巴万星前体b0的纯度、单杂含量、层析收率以及洗脱体积(cv)如下表3所示。表3:洗脱phb0%单杂%收率%实施例18.097.800.0385实施例107.097.090.2179实施例119.095.300.5183由表3可以看出,在实施例1基础上改变洗脱流动相ph条件为7.0,填料对杂质的分离效果降低,洗脱液b0纯度变化不明显,但单杂超出限度。当ph9.0时,b0纯度降低且及单杂明显升高。因此,不同流动相ph影响洗脱结果、杂质分离效果。当洗脱ph8.0时洗脱效果最佳。实施例12聚合物层析对a40926分离纯化结果称取a40926粗品,配置10mg/ml(以b0计)的样品,b0占比82.12%,单杂x01.54%,单杂x0.85%,加入5%甲醇有机相,样品完全溶解后,用氢氧化钠调节样品ph5.0,超声脱气处理。用孔径为0.45μm滤膜处理样品,收集滤液待用。选用unipm30-500聚合物填料(纳微科技有限公司生产),填料粒径30um,孔径500a,装柱5×12cm,柱体积20ml。用0.1mol/l氢氧化钠:乙醇=1:1再生液以3ml/min流速再生聚合物填料5cv,然后用20%乙醇水溶液冲洗填料ph中性。上样前先用ph5.0,甲醇浓度5%的流动相以3ml/min的流速平衡色谱柱5cv,然后用1ml/min的流速上样,上样载量15g/l。上样后用ph5.0甲醇浓度5%的流动相冲洗色谱柱。采用ph5.030mmol/l醋酸钠水溶液和甲醇有机溶剂作为流动相进行梯度洗脱,流速控制在3ml/min。洗脱先用甲醇浓度40%的流动相预洗10cv,分离杂质、降低a组分占比,然后直接用45%的流动相12个柱体积洗脱a40926主峰,收集洗脱液至主峰吸光值下降至峰高2/3,最后用上述氢氧化钠再生液再生填料。洗脱液经hplc高效液相有关物质法分析检测,洗脱液中a40926-b0的纯度91.62%,单杂x0.67%,单杂x01.71%,如附图5,层析收率75%。结果分析:unipm30-500聚合物填料纯化提升b0色谱纯度至91.62%,但对于b0相近的单杂x0及关键单杂x基本无纯化效果。发明人通过优化有机相比例、洗脱ph等条件,虽然对于b0纯度可提升至94%,但对于单杂依旧没有较好的分离效果。因此,粒径30um聚合物微球填料分辨率满足不了工艺需求。所以此工艺中填料选择粒径更细、分辨率更高的c18高分子微球层析介质。在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1