一种辅酶Q10胶囊溶出曲线的测定方法与流程
本发明属于药物分析
技术领域:
,具体涉及辅酶q10胶囊溶出曲线的测定方法。
背景技术:
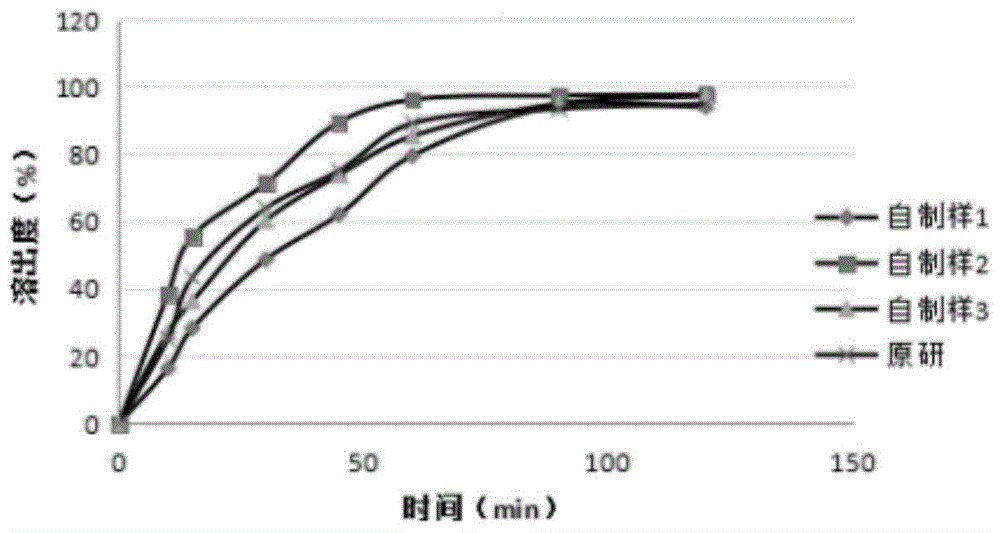
:为全面提升国产仿制药的质量,达到与原研制剂体内、外质量和疗效一致的水平,制定科学合理的固体制剂溶出曲线,是提高体内生物等效性试验成功率的重要步骤,并为将药品特征溶出曲线列入相应的质量标准提供依据,为药品批间质量的一致性、工艺变更前后药品质量的一致性提供保证。就目前国内的合成和工艺水平,杂质控制已经游刃有余,但是要做到体内生物等效还是有一定难度,为了能对体内生物等效性试验做出预判,提高生物等效性试验的成功率,建立一种区分力强的体外溶出检测方法则成为了固体口服制剂一致性评价工作的重点和难点。辅酶q10(coenzymeq10,结构如下式1)又称泛醌,在呼吸链中是一种与蛋白质结合不紧密的辅酶,是生物体内广泛存在的脂溶性醌类化合物,在人体呼吸链中质子移位及电子传递中起重要作用。辅酶q10作为细胞代谢和细胞呼吸激活剂,也是重要的抗氧化剂和非特异性免疫增强剂,能抑制线粒体的过氧化,具有促进氧化磷酸化反应,保护生物膜结构完整性的作用。此外,辅酶q10对免疫非特异性具有增强的作用,可以增强抗体产生的能力,改善t细胞功能。辅酶q10实质上是一种代谢激活剂,能激活细胞呼吸,加速产生三磷酸腺苷(atp),并起到解毒急救的作用;它还可改变细胞和组织的缺氧状态,对肝、脑、心脏及神经系统均有较好的保护和改善作用,增强体内的非特异性免疫应答。经证实,其可以辅助治疗心血管疾病(如:病毒性心肌炎、慢性心功能不全)、肝炎(如:病毒性肝炎、亚急性肝坏死、慢性活动性肝炎)、癌症的综合治疗(能减轻放疗、化疗等引起的某些不良反应)。关于辅酶q10的稳定性及溶解性,段鹏杰等人在文献《辅酶q10的溶解度测定及稳定性考察》中报道了q10的溶解性及稳定性,辅酶q10在氯仿、苯、丙酮、乙醚或石油醚中溶解,在乙醇中极微溶解,在水中不溶;在碱性及光照条件下易被破坏。目前,各国药典中收载的辅酶q10胶囊质量标准中,均未规定溶出度的检查;usp40标准中规定的溶出度检查方法仅限于水溶性辅酶q10,采用桨法操作,转速为每分钟75,以水500ml为溶出介质,经60分钟时取样,限度为标示量的75%。但是本品胶囊在水中几乎不溶,无法评价其与原研制剂间体外溶出行为的差异,为体内生物等效性试验做出预测。技术实现要素:本发明的目的在于克服现有标准的不足,提供一种辅酶q10胶囊溶出曲线的评价方法,具有科学、耐用及可重现等特点,不仅可以用于仿制药与原研药的质量一致性(相似性)评价工作,也可以为药品批间质量的一致性提供保证。为了实现上述发明目的,本发明提供如下技术方案:一种辅酶q10胶囊溶出曲线的测定方法,采用桨法测得辅酶q10胶囊在不同时间点的溶出度,然后绘制成溶出曲线,其中,溶出介质为含有曲拉通和聚山梨酯的混合溶液。在本发明技术方案的建立过程中,考察了不同溶出方法对本品溶出行为的影响。目前,溶出度测定的标准均采用桨法操作,辅酶q10胶囊在溶出介质中会有漂浮的现象,所以,优选以加沉降篮的桨法溶出,即将辅酶q10胶囊装入沉降篮后桨法溶出。对于辅酶q10胶囊的溶出度测定,难点在于溶出介质的筛选。发明人考察了不同溶出介质对辅酶q10胶囊溶出行为的影响。分别以水、5%聚山梨酯80溶液、5%十二烷基硫酸钠溶液、异丙醇-1%聚山梨酯80溶液(25:75)、5%曲拉通x-100溶液为溶出介质进行考察,结果辅酶q10胶囊在聚山梨酯80和十二烷基硫酸钠水溶液中的溶解性较差,几乎不溶。对于异丙醇和聚山梨酯80的介质,溶出6小时仍无法溶解完全,且异丙醇为有机溶剂,不建议作为溶出介质使用。因此采用常规的表面活性剂及有机溶剂不适合本品的溶出度考察。在5%曲拉通x-100溶液中,辅酶q10有一定溶出,但无法完全溶出。但经过进一步试验发现,采用含有曲拉通与聚山梨酯的混合溶液作为溶出介质时,辅酶q10可完全溶出,而且对不同工艺及处方的辅酶q10胶囊有较好的区分力。作为优选,曲拉通的浓度为2-10wt%时,相较于本发明所限定的浓度范围内的其他值,溶出曲线的区分效果进一步得到较大提升,在超出这一范围(尤其是高于10%时),区分效果都会下降。作为优选,聚山梨酯为聚山梨酯80,其浓度为0.1-2wt%。在本发明技术方案的建立过程中,考察了不同转速对本品溶出行为的影响。本品溶解性较差,当转速为每分钟50转和75转时,本品溶出较慢,不适用于溶出行为相似性的比较,所以,本发明将转速设定为100转/分。本发明中,采用桨法检测辅酶q10胶囊的溶出度的方法为:照中国药典2015年版四部通则0931溶出度测定法,以1000ml含有曲拉通和聚山梨酯的混合溶液为溶出介质,转速为每分钟100转,在不同时间点取溶液,滤过,作为供试品溶液,照中国药典2015年版四部通则0512高效液相色谱法,以275nm为检测波长测定并计算辅酶q10胶囊的溶出量。对于取样时间的设置方面,作为优选,在溶出时间0-60min之间设置3-5个时间点取样作为供试品溶液,在60min之后设置2-3个时间点取样作为供试品溶液。进一步地,在所述溶出时间为10、15、30、45、60、90、120min时分别取样作为供试品溶液。采用如上方法进行测定时,所述辅酶q10胶囊在120min时的溶出度大于90%。本发明进一步发现,在测定方法的各影响因素中,下述各因素对其检测效果也有一定程度的影响,进而对其进行了优化,使得整体技术方案的区分效果进一步得到提升。作为优选,所述溶出介质经脱气处理。作为优选,取样后进行过滤,取续滤液进行检测;优选采用0.22μm滤膜过滤。作为优选,采用高效液相色谱法检测溶出度,其中,高效液相的色谱条件为:色谱柱:采用watersmicrobondapakc18色谱柱流动相:甲醇:无水乙醇=65:35流速:1.0ml/min进样体积:20μl。进一步地,所述洗脱程序为等度洗脱。本发明还提供了一种对辅酶q10胶囊溶出曲线的相似性进行评价的方法,其中,采用相似因子法,利用参比制剂与供试制剂在不同时间点测得的平均溶出度数据,计算相似因子f2,所述相似因子f2用于评价参比制剂与供试制剂之间的相似性。与现有技术相比,本发明所取得的有益效果是:(1)本发明中的方法,解决了现有技术中常规的溶出度测定方法无法准确衡量辅酶q10胶囊的溶出行为的技术问题,能更好的反映辅酶q10胶囊的质量,使得溶出度的测定结果与体内溶出效果更接近,更具有实际参考意义。(2)该方法具有科学、耐用及可重现等特点,可以有效区分不同处方和工艺变量对辅酶q10的体外释放的影响,不仅可以用于仿制药与原研药的质量一致性评价工作,也可以为药品批间质量的一致性提供保证,进而保证药品的质量,达到质量与疗效的一致性。附图说明图1为本发明测定方法测定的三批自制样品与一批原研的溶出曲线图2为本发明测定方法测定的六粒原研样品的溶出曲线图3为本发明测定方法重复测定三次的自制样品溶出曲线。具体实施方式下面通过具体实施例对本发明的方法进行说明,以使本发明技术方案更易于理解、掌握,但本发明并不局限于此。下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。溶出度方法学研究1溶出介质的选择1.1溶出介质的制备称取曲拉通x-100、聚山梨酸酯80各适量,加水溶解后,定容至1000ml,摇匀即得。1.2色谱条件:色谱柱:采用watersmicrobondapakc18(4.6mm×150mm,10μm)色谱柱流动相:甲醇:无水乙醇=65:35检测波长:275nm流速:1.0ml/min进样体积:20μl;柱温:30℃;等度洗脱。1.3溶出曲线测定方法取辅酶q10胶囊装入沉降篮后,采用桨法(桨法参照药典2015年版四部通则0931溶出度测定法),分别以混合溶液a(5wt%曲拉通、水)、b(5wt%曲拉通+0.1wt%聚山梨酯80水溶液)、c(5wt%曲拉通+0.2wt%聚山梨酯80水溶液)、d(5wt%曲拉通+0.5wt%聚山梨酯80水溶液)、e(5wt%曲拉通+1wt%聚山梨酯80水溶液)和f(5wt%曲拉通+2wt%聚山梨酯80水溶液)为溶出介质,转速为每分钟100转,经10分钟、15分钟、30分钟、45分钟、60分钟、90分钟、120分钟时,分别取溶液2ml,滤过,作为供试品溶液,并即时补充相同温度、相同体积的溶出介质;分别取各时间点的溶液,照中国药典2015年版四部通则0512高效液相色谱法,测定并计算辅酶q10胶囊的累计溶出率(%),结果如下表1。表1辅酶q10胶囊在不同溶出介质中的累计溶出率结果由表1数据可以看出:仅以曲拉通溶液为溶出介质时,辅酶q10胶囊120min时的累计溶出率小于65%,当以含有曲拉通和聚山梨酯80的混合溶液体系为溶出介质时,辅酶q10胶囊120min时的累计溶出率均大于90%。如下将以混合溶液e为例,进一步考察其溶出行为。实施例1一种辅酶q10胶囊的溶出曲线测定方法,该方法具体包括如下步骤:溶出介质:混合溶液e(5wt%曲拉通+1wt%聚山梨酯80水溶液)。采用1.3的溶出度测定方法对不同处方(表2)的三批自制样品及原研样品进行溶出曲线测定,不同处方之间溶出行为的差异明显,说明本法具有较强的区分效力,适用于该胶囊溶出曲线的测定,具体的测定结果如下表3,溶出曲线见附图1。表2样品信息批号自制样品1q-20190601自制样品2q-20190703自制样品3q-20190905原研制剂85a77s表3辅酶q10胶囊的溶出曲线测定结果采用本法对一批原研样品进行溶出曲线测定,六粒样品在不同时间点的累计溶出率的相对标准偏差均较好(小于10%),说明本法的均一性良好,适用于该胶囊溶出曲线的测定,具体的测定结果如下表4,溶出曲线见附图2。表4同一批次原研样品平行六粒的溶出曲线测定结果原研0min10min15min30min45min60min90min120min1024.7542.9261.6675.8084.7891.4495.472025.9643.8965.3773.4892.4794.4793.523028.1445.5565.8377.1590.6596.1496.544022.6242.5163.1675.5885.4791.8495.445024.1443.9064.3871.0590.8291.4693.526025.1941.2062.8377.5090.2795.4994.42均值025.1343.3363.8775.0989.0893.4794.82rsd%/7.373.412.513.253.552.291.27采用本法对同一批自制样品重复测定三次,三次测定结果无明显差异,说明本法的重复性良好,适用于该胶囊溶出曲线的测定,测定结果如下表5,溶出曲线见附图3。表5同一批次样品重复三次的溶出曲线测定结果自制样30min10min15min30min45min60min90min120min第一次026.3538.2858.6572.0679.5194.3797.03第二次023.6235.6157.4970.5678.8992.8495.09第三次027.8039.3460.6772.9682.5493.6596.86综合以上结果,本法具有较强的区分能力,可区分出不同处方之间的差异,且均一性和重复性均符合要求,说明本方法适用于评价该胶囊批次间的差异,同时也可以评价其与原研制剂间体外溶出行为的差异,为体内生物等效性试验做出预测,提高生物等效性试验的成功率。最后应当说明的是,以上内容仅用以说明本发明的技术方案,而非对本发明保护范围的限制,本领域的普通技术人员对本发明的技术方案进行的简单修改或者等同替换,均不脱离本发明技术方案的实质和范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1