一种海藻中氨基糖、中性糖和糖醛酸同时快速检测的方法与流程
本发明属于食品工业中糖类化合物的分析检测技术领域,具体涉及一种海藻中氨基糖、中性糖和糖醛酸同时快速检测的方法。
背景技术:
当今世界已经进入资源环境瓶颈期,开发海洋资源为可持续发展的战略选择。海洋生物相比于陆地生物具有独特的优势。我国海洋资源丰富,海藻加工及其高值化利用成为我国海洋资源综合利用的重要领域。海藻多糖在降血脂、抗氧化、抗凝血、抗肿瘤、抗疲劳、抗炎、免疫调节和神经保护作用等方面具有显著效用,在生物化学、保健食品及临床医学等领域的应用也越来越受到重视。在欧美、日本等国家,褐藻糖胶已作为保健品在市场上广泛流通;近年来,国内市场也涌现出多种形式的褐藻糖胶产品。但目前关于海藻多糖的单糖和寡糖组成、结构特征等尚未明确,影响了海藻多糖产品的开发和市场推广。海藻多糖的生物学活性与其糖类组成密不可分。多糖的生物活性与其糖类组成密切相关,因此研究多糖的糖类组成对研究其生物活性有重要意义。海藻多糖中单糖种类繁多,定性定量工作极具挑战。单糖是一种含有多羟基的亲水性大分子,为多羟基醛、酮及其衍生物,由于其极性大、无紫外和荧光基团、结构相似,很多互为同分异构体,所以海藻多糖的单糖分离及定量分析极具挑战,成为制约糖类生物活性研究及应用的瓶颈。
目前,糖类的检测方法主要有:液相色谱法、气相色谱法、毛细管电泳法和离子色谱法等。液相色谱检测糖类化合物分为两种模式:非衍生化的直接检测法和衍生化后的检测法。各种糖类检测方法之间的比较详见表1。直接检测的液相色谱法操作简便,但检测灵敏度低、重现性较差;衍生化的液相色谱法提高了检测灵敏度和重现性,但是衍生过程操作繁琐,耗时,且在衍生过程会不可避免的造成误差,不适用于常规的食品工业中糖类检测的应用。气相色谱法同样需要对糖类化合物进行衍生化处理,衍生化操作常需要多种试剂,操作繁琐,定量的重复性差。毛细管电泳利用极细内径的色谱柱来分离单糖,在复杂的单糖的分离度具有独特优势,但由于其色谱柱极细的内径和极低的样品注射量,导致检测灵敏度较低,对于含量较少的样品的检测具有不适用。离子色谱法检测单糖具有灵敏度高、分离度高、前处理操作简便、样品用量少等优点,适用于常规食品工业中的单糖检测,但是hpaec在色谱系统稳定性方面一直存在问题。
表1糖类不同检测方法的比较
针对于hpaec-pad方法,现有技术中“定量测定褐藻(海带科)碳水化合物成分的方法”(methodologyforquantitativedeterminationofthecarbohydratecompositionofbrownseaweeds(laminariaceae),doi:10.1039/c4ra03537b(paper)rscadv.,2014,4,25736-25746)公开了海带中中性糖、糖醛酸总含量、灰分、水分的测定;其针对的糖类为:岩藻糖、葡萄糖、甘露醇、古罗糖醛酸、葡萄糖醛酸、半乳糖醛酸等6种;并采用两步水解法进行样品的前处理;该文献中关于糖类的检测侧重于中性糖和糖醛酸总量的测定,而且没有糖类检测的相关方法学验证的报道。海藻中含有大量的蛋白质,蛋白质的存在污染色谱柱,影响分离效果,降低柱效,该文献的样品前处理并未对蛋白杂质进行去除;此外,该文献中检测的糖的种类也较少;因而,亟需一种同时检测多种糖类、无需衍生化处理、检测结果精确、稳定性高的糖类检测方法。
技术实现要素:
糖类化合物的检测在业内一直是一项具有挑战性的工作,中性糖、氨基糖和糖醛酸的同步分离更具难度。现有技术存在衍生化操作繁琐,耗时、分离度低、灵敏度低,重现性差、线性范围小等缺点。由于糖类其强极性、无发色基团和荧光基团,在使用常规检测器(如紫外检测器,荧光检测器等)常需对糖类进行衍生化,衍生化过程常常伴随着操作繁琐、耗时,不可避免会引入误差等缺点。
针对现有技术中存在的问题,本发明的目的在于提供一种海藻中氨基糖、中性糖和糖醛酸同时快速检测的方法;利用离子色谱-脉冲安培检测法解决中性糖、氨基糖和糖醛酸多组分同时分析的技术问题。本发明为海藻(海带、紫菜、裙带菜等)中海藻多糖的单糖组成的检测,建立了一种快速、简便、灵敏度高、加标回收率高、稳定性好、重现性好,适用于常规食品工业中糖类化合物的分析检测技术,可实现快速分析和绿色分析。
为了达到上述目的,本发明采用如下技术方案:
海藻中氨基糖、中性糖和糖醛酸同时快速检测的方法,步骤如下:
待测样品采用超声辅助水提醇沉的方法提取海藻多糖粗品;利用sevage法去除提取的海藻多糖粗品中的蛋白后,采用三氟乙酸水解海藻多糖;水解完成后加入naoh中和水解液的酸度,过0.22μm滤膜后用于离子色谱-脉冲安培检测器检测,利用保留时间对待测样品中的糖类进行定性,将定性后糖类的峰面积导入该糖类对应的标准工作曲线中,计算获得该糖类的浓度,从而实现定量分析。
在上述方案的基础上,所述离子色谱-脉冲安培检测的检测条件为:
色谱柱:dionexcarbopacpa10(250mm×4mm)糖分析专用柱;
dionexcarbopacpa10(50mm×4mm)糖保护柱;
工作电极:gold;
参比电极:agcl电极;
工作电位:糖标准四电位,具体如下:
流速:0.4~1.2ml/min;
柱温:20~40℃;
进样量:25μl;
采用多级梯度洗脱程序进行洗脱;
所述洗脱程序中的a为超纯水,b为200mmnaoh,c为500mmnaac;
所述离子色谱-脉冲安培检测过程中采用多级梯度洗脱程序;具体如下:
*curve5是线性;上述溶液均使用电阻率为18.2mω·cm以上的超纯水配置。
在上述方案的基础上,所述流速为1.0ml/min。
在上述方案的基础上,所述柱温:25℃。
在上述方案的基础上,所述中性糖为岩藻糖、葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖中的至少一种;所属氨基糖为氨基半乳糖、氨基葡萄糖中的至少一种;所述糖醛酸为半乳糖醛酸、葡萄糖醛酸中的至少一种。
在上述方案的基础上,所述标准工作曲线是由以下方法制备而成:
配置岩藻糖、葡萄糖、氨基半乳糖、氨基葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖、半乳糖醛酸和葡萄糖醛酸的混合标准工作溶液,混合标准工作溶液浓度梯度分别为0.5mg/l、2mg/l、5mg/l、10mg/l、20mg/l、40mg/l、60mg/l;
上述混合标准溶液分别用于离子色谱-脉冲安培检测器检测,以标准溶液中岩藻糖、葡萄糖、氨基半乳糖、氨基葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖、半乳糖醛酸和葡萄糖醛酸的浓度为横坐标,再以标准溶液中岩藻糖、葡萄糖、氨基半乳糖、氨基葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖、半乳糖醛酸和葡萄糖醛酸在脉冲安培检测器上的响应值的峰面积为纵坐标绘制标准工作曲线。
在上述方案的基础上,所述的海藻为海带、紫菜、裙带菜中的至少一种。
上述海藻中氨基糖、中性糖和糖醛酸同时快速检测的方法在食品工业中糖类化合物分析检测中的应用。
本发明技术方案的优点:
本发明检测结果准确,稳定性好,检出限低,为1.27~47.49μg/l,检测的所有糖类化合物均达到基线分离的效果,线性相关系数较好(r>0.998),日内精度<1.69%,日间精度<1.78%,线性范围在0.5~60mg/l之间。
本发明前处理方法简单快速,无需衍生,大幅提高方法的适用性、回收率。本发明利用超声破碎的原理来提取海藻中的海藻多糖,可加快胞内多糖的溶出,提高提取效率。后经去除蛋白杂质过程后,三氟乙酸水解多糖,便可直接进样。省去常规方法对糖类进行衍生化的操作,避免了衍生化过程引入不必要的误差。相对于衍生化-液相色谱法和气相色谱法等检测方法,本发明前处理更为简便,易于操作,节约时间,适合常规食品工业的糖类的检测。
本发明的方法采用的离子色谱技术利用糖类在强碱性条件下,呈现阴离子状态,从而经过糖分析柱达到分离的效果,无需衍生,有效规避了衍生化这一过程。
由于海洋类生物中糖的种类多种多样,其多组分物质同时检测,一直是分析化学领域的技术难点。本发明解决了目前采用多种检测技术、多种仪器、多种前处理方法进行多种组分测定时效率低的技术难题,大幅提高了检测效率,降低了检测成本。
现有技术中离子色谱法存在重现性差、仪器不稳定等弱点。本发明通过在采用500mmol/l醋酸钠溶液清洗色谱柱10min达到基质消除的结果后,采用200mmol/l氢氧化钠溶液作为赋予系统强ph值的条件,使检测方法的重现性显著提高。
目前关于水产品中糖类检测技术还较为缺乏,本发明操作简便、快速,适用于常规检测的应用,为水产品中糖类组成的检测提供依据。
附图说明
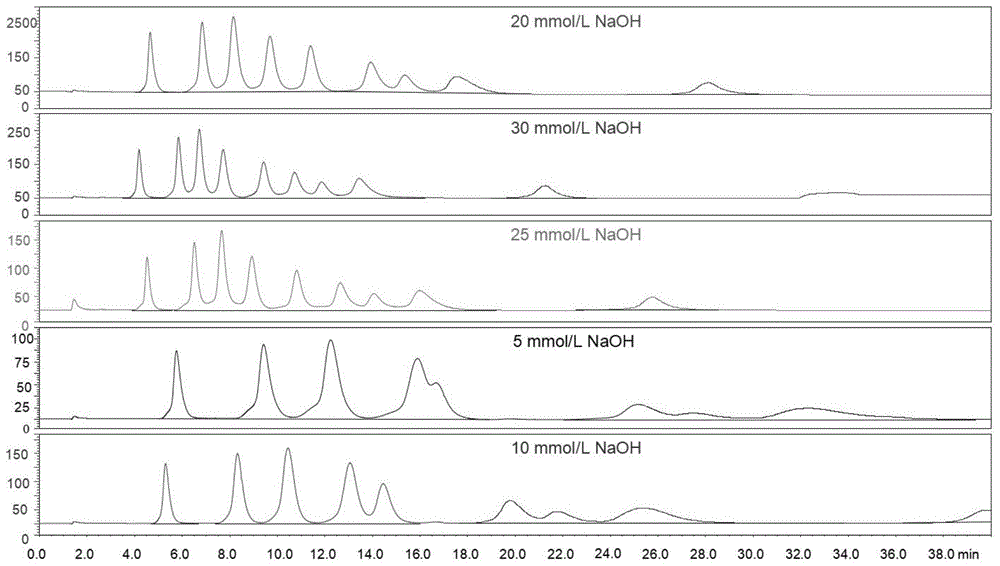
图1为naoh浓度对糖类色谱图的影响,其中,横坐标为糖类化合物在色谱柱中的保留时间(单位:min),纵坐标为糖类物质在脉冲安培检测器上的响应值(单位为:nc);
图2为体积流量对糖类色谱图的影响,其中,横坐标为糖类化合物在色谱柱中的保留时间(单位:min),纵坐标为糖类物质在脉冲安培检测器上的响应值(单位为:nc);
图3为柱温对糖类色谱图的影响,其中,横坐标为糖类化合物在色谱柱中的保留时间(单位:min),纵坐标为糖类物质在脉冲安培检测器上的响应值(单位为:nc);
图4混合标准溶液色谱图,其中,横坐标为糖类化合物在色谱柱中的保留时间(单位:min),纵坐标为糖类物质在脉冲安培检测器上的响应值(单位为:nc)。
具体实施方式
在本发明中所使用的术语,除非有另外说明,一般具有本领域普通技术人员通常理解的含义。
下面结合具体实施例,并参照数据进一步详细的描述本发明。以下实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围
实施例1
海藻中氨基糖、中性糖和糖醛酸同时快速检测的方法,主要包括以下步骤:
(1)海带中海带多糖的提取:先将新鲜采摘的海带洗净表面的泥沙、盐渍等杂质,在50℃烘箱中烘干,用粉碎机打碎成海带粉,过60目网筛,-20℃保存备用。取5g海带粉用10倍体积的无水乙醇除去色素、油脂等杂质,在50℃干燥箱中干燥至恒重。加入30倍超纯水,利用超声细胞粉碎机处理30min,置于60℃恒温水浴锅中浸提3h,离心取上清液。于60℃旋转蒸发器浓缩至原体积的1/4,加无水乙醇至质量分数为80%,4℃过夜。离心,取沉淀复溶于水,冻干后即得海带多糖粗品。
(2)除蛋白:取步骤(1)中20mg海带多糖粗品溶于4ml水中,加入1mlsevage溶液(正丁醇:氯仿=1:5),振荡混匀20min,小心吸取上层水相,重复上述操作,直至无蛋白析出,得多糖溶液。
(3)海带多糖的水解:取步骤(2)中多糖溶液加入同等体积的4mol/l三氟乙酸溶液,于120℃恒温干燥箱中水解2h;水解完成后naoh中和水解液,离心,去沉淀,经0.22μm滤膜过滤,用于离子色谱分析。
(4)混合标准溶液的配置:配置岩藻糖(fuc)、葡萄糖(glc)、氨基半乳糖(galn)、氨基葡萄糖(glcn)、半乳糖(gal)、甘露糖(man)、果糖(fruc)、核糖(rib)、乳糖(lac)、半乳糖醛酸(galua)和葡萄糖醛酸(glcua)的混合标准工作溶液,混合标准工作溶液浓度梯度分别为0.5mg/l、2mg/l、5mg/l、10mg/l、20mg/l、40mg/l、60mg/l。
(5)待测样品的检测:分别将步骤(3)中的待测样品溶液和步骤(4)中的混合标准溶液分别用于离子色谱-脉冲安培检测器检测,获得以标准溶液中岩藻糖、葡萄糖、氨基半乳糖、氨基葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖、半乳糖醛酸和葡萄糖醛酸的浓度为横坐标,再以标准溶液中岩藻糖、葡萄糖、氨基半乳糖、氨基葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖、半乳糖醛酸和葡萄糖醛酸在脉冲安培检测器上的响应值的峰面积为纵坐标绘制标准工作曲线,利用各糖类化合物在横坐标上的保留时间来对样品中的糖类进行定性,利用工作曲线计算获得待测样品溶液中岩藻糖、葡萄糖、氨基半乳糖、氨基葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖、半乳糖醛酸和葡萄糖醛酸的浓度从而定量。
上述步骤(5)离子色谱-脉冲安培检测的检测条件为:
色谱柱:dionexcarbopacpa10(250mm×4mm)糖分析专用柱;
dionexcarbopacpa10(50mm×4mm)糖保护柱;
工作电极:gold;
参比电极:agcl电极;
工作电位:糖标准四电位,具体如下:
表2糖标准四电位波形
流速:1.0ml/min;
柱温:25℃;
进样量:25μl;
采用多级梯度洗脱程序进行洗脱;
所述洗脱程序中的a为超纯水,b为200mmnaoh,c为500mmnaac;所述离子色谱-脉冲安培检测过程中采用多级梯度洗脱程序;具体如下:
表3多级梯度洗脱程序
*curve5是线性;上述溶液均使用电阻率为18.2mω·cm以上的超纯水配置。
实施例2
淋洗相洗脱系统对检测结果的影响
(1)不同氢氧化钠浓度对糖类色谱图的影响
分别以5、10、20、25、30mmol/lnaoh溶液对9种糖类化合物(fuc、glc、galn、glcn、gal、man、fruc、rib和lac)进行等度洗脱,检测不同氢氧化钠浓度对色谱峰的分离度与峰形的影响,结果如图1所示。试验过程中发现:随naoh浓度的增大,各个色谱峰保留时间缩短,响应值增高,这可能是由于随naoh浓度增大,单糖解离度增大。这是由于淋洗液naoh溶液浓度分离出来的oh-可以从两个方向上对单糖分离产生影响,一方面,随naoh浓度增大,oh-浓度增大,单糖解离度增大,糖类在固定相的保留增强,导致色谱峰保留时间延长;另一方面,随naoh浓度增大,oh-浓度增大,导致淋洗能力增强,导致色谱峰保留时间缩短。其中第二方面占据主导优势。当浓度为5mmol/lnaoh溶液时,色谱峰展宽,分析时间较长,且洗脱时间超过60min;10mmol/lnaoh溶液时,fuc、glc、galn三种单糖之间的分离度较高,峰形较好,但洗脱时间超过40min。当淋洗液naoh浓度大于20mmol/l、时,随着naoh浓度增大,各单糖之间的分离度呈现下降的趋势。其中20mmol/lnaoh能够在保证分离度的情况下,又能消除滞留时间长的单糖的拖尾现象,节省分析时间,且分离度达到实验要求。
(2)不同体积流量对糖类色谱图的影响
以20mmol/lnaoh溶液作为淋洗液,分别设定淋洗液的体积流量为0.4、0.6、1.0、1.2ml/min,检测不同体积流量对9种糖类化合物(fuc、glc、galn、glcn、gal、man、fruc、rib和lac)的色谱峰的分离度与峰形的影响,结果如图2所示。结果表明,体积流量低时分析时间长,峰形变宽,柱效降低。体积流量低时,对于易洗脱的单糖(fuc、glc、galn、glcn和gal)分离度更高,峰形更好;体积流量的变化对于man、fruc、rib和lac等较难洗脱的糖类则影响较小;体积流量高时,柱压升高,各糖类分离度变差。考虑分离度、柱压、柱效及分析时长等因素,1.0ml/min的体积流量效果最好。
(3)不同柱温对糖类色谱图的影响
柱温对hpaec分离单糖具有重要的影响,很小的温差变化(±5℃)便会引起单糖的保留时间的偏移、以及单糖保留时间复现性。糖类在色谱柱上的保留属于放热行为,符合范托霍夫方程,随温度增大,糖类的解离度增大。不同糖类受温度影响程度不同,一般来说,糖类的相对分子质量越大,保留时间随温度变化的程度就越大。以20mmol/lnaoh溶液作为淋洗液,流速为1.0ml/min,改变色谱柱温度20、25、30、35、40℃,观察待测组分色谱峰分离度以及峰形的变化发现,结果如图3所示。随温度增加,色谱峰的保留时间呈减小的趋势;当色谱柱温度在30℃以上时,糖的分离度变差,不能实现葡萄糖和氨基半乳糖的分离;当色谱柱温度低于25℃时,各色谱峰变宽,洗脱时间太长。综合考虑到分离度和峰形以及仪器耐受性,以25℃作为色谱柱温度最佳。
实施例3
(1)本发明检测方法的线性关系与检出限
检测实施例1步骤4配置的不同浓度的混合标准工作溶液,计算线性方程及其相关系数,利用色谱峰信噪比来计算待测组分的检出限(s/n=3),定量限(s/n=10)。线性相关系数(r)大于0.998,检出限在1.27~47.49μg/l之间。
表4糖类检出限以及线性数据
(2)样品加标回收率
对11种糖类化合物添加适当含量的加标量,重复进样7次,计算峰面积的相对标准偏差(rsd%)和加标回收率,加标回收率在89.35~115.67%之间。
表5不同加标浓度下样品的回收率及相对标准偏差(n=7)
nd:notdetected
(3)精密度
精密度从日内精度和日间精度两个方面来考察。质量浓度为10mg/l的混合标准溶液连续进样7次计算器日内精度,各单糖保留时间的rsd均小于0.25%,峰面积的rsd均小于1.69%。将10mg/l的混合标准溶液一天进样3次,连续进样4天,计算其日间精度,各单糖保留时间的rsd均小于0.27%,峰面积的rsd均小于1.78%。
表6精密度实验结果
综上所述,本发明利用离子色谱-脉冲积分安培检测器,糖标准四电位作为检测电位,色谱柱carbopacpa10(250mm×4mm)作为糖分析柱,carbopacpa10(50mm×4mm)作为糖保护柱,柱温为25℃,流速为1.0ml/min,采用水-naoh-naac组成三元淋洗液,多级梯度淋洗,同时检测岩藻糖、葡萄糖、氨基半乳糖、氨基葡萄糖、半乳糖、甘露糖、果糖、核糖、乳糖、半乳糖醛酸、葡萄糖醛酸11种糖类化合物。
本发明前处理方法操作简便,利用超声破碎的原理,可加快胞内多糖的溶出,提高提取效率。经取蛋白过程后,利用三氟乙酸将多糖水解成单糖,便可直接进样。省去常规方法对糖类进行衍生化的操作,避免了衍生化过程引入不必要的误差。
结果:测得各糖类化合物其相应范围内的线性关系良好(r>0.998);其检出限为1.27~47.49μg/l,灵敏度高;日内精度和日间精度为1.35%~2.80%,重现性和复现性高;样品加样回收率为89.35%~115.67%,加标回收率高。
图4为10mg/l的11种混合标准溶液按实施例1步骤5中的色谱条件进样所得出的色谱图。由该图得知,本发明中的11中糖类化合物在该方法条件下,达到基线分离,峰形好,响应值高,灵敏度高。
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!