一种一清颗粒指纹图谱质量控制方法与流程
本发明属于指纹图谱分析
技术领域:
,特别涉及一种一清颗粒指纹图谱质量控制方法。
背景技术:
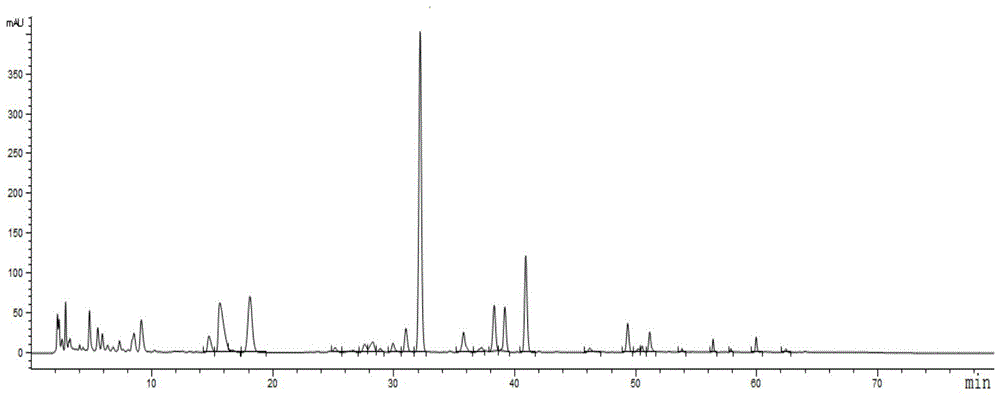
:一清颗粒由黄连、大黄和黄芩3味中药制成,具有清热泻火解毒,化瘀凉血止血的功效。用于火毒血热所致的身热烦躁、目赤口疮、咽喉牙龈肿痛、大便秘结、吐血、咯血、衄血、痔血;咽炎、扁桃体炎、牙龈炎见上述征候者。一清颗粒的现行质量标准中,采用薄层色谱法分别对三味药材及主要组分进行鉴别,采用高效液相色谱法测定了黄芩苷的含量。中药多成分的复杂性决定了单一成分难以准确表达中药的内在质量,指纹图谱能全面反映中药所含成分,可从整体上表征中药产品的质量。因此,为使该制剂的质量控制手段更严密和科学,提供一种一清颗粒指纹图谱质量控制方法,能更加全面有效地控制一清颗粒的质量,为进一步探索和研究制剂的质量提供科学依据。技术实现要素:本发明的目的是针对现有技术的不足,提供一种一清颗粒指纹图谱质量控制方法。本发明的目的是通过以下技术方案实现的:一种一清颗粒指纹图谱质量控制方法,具体步骤为:(1)制备20批次以上的待测产品溶液,每批次待测产品溶液的制备过程如下:取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入70vol%甲醇50ml,称重,加热回流或超声处理30-60min,放冷,用70vol%甲醇补足减失的重量,摇匀,离心,取上清液,即得该批次待测产品溶液;(2)参照物溶液的制备:取黄芩苷对照品适量,加甲醇制成每1ml含50μg的溶液,即得。(3)采用高效液相色谱法,以浓度为0.2vol%磷酸溶液a、甲醇b、乙腈d为流动相,分别获取所有批次的待测产品溶液和参照物溶液的高效液相指纹图谱;色谱条件为:采用welchlp-c18色谱柱,色谱柱长度为250mm,色谱柱内径为4.6mm,填料粒径为5μm;线性梯度洗脱程序为:0~22min内,流动相中a的体积分数为76%,b的体积分数为1%,d的体积分数为23%;22~48min内,流动相中a的体积分数为76%~40%,b的体积分数为1%~50%,d的体积分数为23%~10%;48~65min内,流动相中a的体积分数为40%~12%,b的体积分数为50%~33%,d的体积分数为10%~55%;65~68min内,流动相中a的体积分数为12%,b的体积分数为33%,d的体积分数为55%;68~72min内,流动相中a的体积分数为12%~76%,b的体积分数为33%~1%,d的体积分数为55%~23%;流动相流速为1.0ml/min;柱温为30℃;进样量为10μl;检测波长为225nm;(4)质量稳定性评价:将步骤(3)得到的参照物溶液和所有批次的待测产品溶液的色谱图,导入中药色谱指纹图谱相似度评价系统,经多点校正后进行mark峰匹配,用平均数法生成对照指纹图谱,并计算相似度,相似度≥0.90的样品评价为质量稳定的产品。进一步地,所述步骤4中,平均数法采用的时间窗为0.1min。本发明的有益效果是:该方法为一清颗粒的内部质量控制提供了一种简单、有效的方法,可使一清颗粒质量的有效性、稳定性、均一性得到科学评价。附图说明图1为260nm波长下的一清颗粒样品色谱图;图2为254nm波长下的一清颗粒样品色谱图;图3为225nm波长下的一清颗粒样品色谱图;图4为不同流动相条件下的一清颗粒样品色谱图;图5为不同梯度洗脱程序条件下的一清颗粒样品色谱图;图6为不同提取方法条件下的一清颗粒样品色谱图;图7为不同提取溶剂条件下的一清颗粒样品色谱图;图8为不同提取时间条件下的一清颗粒样品色谱图;图9为连续进样6次获取同一供试品溶液的指纹图谱;图10为重复6次获取同一批次的供试品溶液的指纹图谱;图11为同一供试品溶液放置不同时间后的指纹图谱;图12为本发明测得的20批次的浙江凯润药业股份有限公司的一清颗粒指纹图谱;图13为本发明测得的浙江凯润药业股份有限公司的一清颗粒对照指纹图谱。具体实施方式本发明提供了一种一清颗粒指纹图谱质量控制方法,具体包括如下步骤:(1)制备20以上批次的待测产品溶液,每批次待测产品溶液的制备过程如下:取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入70vol%甲醇50ml,称重,加热回流或超声处理30-60min,放冷,用70vol%甲醇补足减失的重量,摇匀,离心,取上清液,即得该批次待测产品溶液。(2)参照物溶液的制备:取黄芩苷对照品适量,加甲醇制成每1ml含50μg的溶液,即得。(3)采用高效液相色谱法,以浓度为0.2vol%磷酸溶液a、甲醇b、乙腈d为流动相,分别获取所有批次的待测产品溶液和参照物溶液的高效液相指纹图谱;色谱条件为:采用welchlp-c18色谱柱,色谱柱长度为250mm,色谱柱内径为4.6mm,填料粒径为5μm;线性梯度洗脱程序为:0~22min内,流动相中a的体积分数为76%,b的体积分数为1%,d的体积分数为23%;22~48min内,流动相中a的体积分数为76%~40%,b的体积分数为1%~50%,d的体积分数为23%~10%;48~65min内,流动相中a的体积分数为40%~12%,b的体积分数为50%~33%,d的体积分数为10%~55%;65~68min内,流动相中a的体积分数为12%,b的体积分数为33%,d的体积分数为55%;68~72min内,流动相中a的体积分数为12%~76%,b的体积分数为33%~1%,d的体积分数为55%~23%;流动相流速为1.0ml/min;柱温为30℃;进样量为10μl;检测波长为225nm。(4)质量稳定性评价:将步骤(3)得到的参照物溶液和所有批次的待测产品溶液的指纹图谱,导入中药色谱指纹图谱相似度评价系统,经多点校正后进行mark峰匹配,用平均数法生成对照指纹图谱,并计算相似度,相似度≥0.90的样品评价为质量稳定的产品。其中,平均数法采用的时间窗为0.1min。下面,结合具体实施例和附图说明对本发明作进一步的说明。以下实施例使用的试剂和仪器如下,但不限于此:1、试剂甲醇、乙腈(色谱纯,美国tedia),水为超纯水,甲醇、乙醇(分析纯,国药集团化学试剂有限公司)。黄芩苷(批号110715-201619)来源于中国食品药品检定研究院。20批一清颗粒由浙江凯润药业股份有限公司提供,批号:1808151、1808161、1808171、180919、180920、180924、180925、180926、190513、190514、190515、190519、190520、190521、190611、190612、190613、190806、190807、190808。2、仪器mettler-toledoxs105电子天平(瑞士mettler公司);agilent1100高效液相色谱仪(美国agilent)。实施例1:检测波长选择考虑到一清颗粒为复方制剂,成分复杂,检测波长的选择应兼顾各类化学组分,经多波长扫描及对比分析,考察了260、254、225nm波长下的一清颗粒样品色谱图,结果如图1~图3所示,以上波长下的色谱峰数无明显差异,225nm波长下的色谱图色谱峰表观丰度较高。因此选择225nm为一清颗粒高效液相指纹图谱检测波长。实施例2:流动相体系选择比较了乙腈-0.2vol%磷酸溶液、甲醇-乙腈-0.2vol%磷酸溶液两种流动相体系,所得色谱图见图4所示。结果甲醇-乙腈-0.2vol%磷酸溶液体系下的色谱图峰分离较好,选择甲醇-乙腈-0.2vol%磷酸溶液体系为一清颗粒色谱分离用流动相体系。实施例3:流动相梯度选择采用welchlp-c18(250mm×4.6mm,5μm)色谱柱;流速:1.0ml/min;检测波长:225nm;柱温:30℃;进样量:10μl;考察了多种梯度洗脱条件,其中的四个线性梯度(梯度1~梯度4)条件如下:梯度1、梯度2以0.2vol%磷酸溶液a、乙腈-甲醇(3:1)b为流动相;梯度1:0~20min:流动相中a体积分数为70%;20~55min:流动相中a体积分数为70%~40%;55~60min:流动相中a体积分数为40%;60~65min:流动相中a体积分数为40%~70%;梯度2:0~20min:流动相中a体积分数为72%;20~30min:流动相中a体积分数为72%~63%;30~35min:流动相中a体积分数为63%;35~60min:流动相中a体积分数为63%~27%60~70min:流动相中a体积分数为27%;70~75min:流动相中a体积分数为27%~72%;梯度3、梯度4以0.2vol%磷酸溶液a、甲醇b、乙腈d为流动相;梯度3:0~22min:流动相中a体积分数为74%;流动相中b体积分数为5%;22~48min:流动相中a体积分数为74%~40%;流动相中b体积分数为5%~50%;48~65min:流动相中a体积分数为40%~12%;流动相中b体积分数为50%~33%;65~68min:流动相中a体积分数为12%;流动相中b体积分数为33%;68~72min:流动相中a体积分数12%~74%;流动相中b体积分数为33%~5%;梯度4:0~22min:流动相中a体积分数为76%;流动相中b体积分数为1%;22~48min:流动相中a体积分数为76%~40%;流动相中b体积分数为1%~50%;48~65min:流动相中a体积分数为40%~12%;流动相中b体积分数为50%~33%;65~68min:流动相中a体积分数为12%;流动相中b体积分数为33%;68~72min:流动相中a体积分数12%~76%;流动相中b体积分数为33%~1%;梯度1-梯度4条件下得到的图谱如图5所示,从图中可以看出,以梯度4得到的色谱图峰分离较好,而且谱峰较多,可给出较大的信息量,故选定以梯度4作为一清颗粒指纹图谱分析的色谱条件。实施例5:提取方法选择取同一批次一清颗粒样品,通过以下方法进行提取:方法(1):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加甲醇50ml,称重,加热回流提取1小时;方法(2):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加甲醇50ml,称重,超声提取1小时;以上样品溶液,放冷,用甲醇补足减失的重量,摇匀,离心,取上清液,注入高效液相色谱仪,记录色谱图,得到的图谱如图6所示。结果,加热回流1小时和超声提取1小时两种提取方法无明显差异,最后选择操作简单的超声提取方法。实施例6:提取溶剂选择取同一批次一清颗粒样品,通过以下方法进行提取:方法(1):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称重,超声处理1小时,放冷,用甲醇补足减失的重量,摇匀,离心,取上清液,即得;方法(2):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入乙醇50ml,称重,超声处理1小时,放冷,用乙醇补足减失的重量,摇匀,离心,取上清液,即得;方法(3):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声处理1小时,放冷,用70%甲醇补足减失的重量,摇匀,离心,取上清液,即得;方法(4):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%乙醇50ml,称重,超声处理1小时,放冷,用70%乙醇补足减失的重量,摇匀,离心,取上清液,即得;以上样品溶液,分别注入高效液相色谱仪,记录色谱图,得到的图谱如图7所示。结果,用70%甲醇作为提取溶剂时提取效果和峰形均较好,最终选择70%甲醇作为提取溶剂。实施例7:提取时间选择取同一批次一清颗粒样品,通过以下方法进行提取:方法(1):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声处理1小时;方法(2):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声处理30min;方法(3):取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声提取45min;以上样品溶液,放冷,用70%甲醇补足减失的重量,摇匀,离心,取上清液,注入高效液相色谱仪,记录色谱图,得到的图谱如图8所示。结果,不同提取时间下结果无明显差异,最终选择提取时间为30min。实施例8:指纹图谱方法学考察1、精密度试验(1)取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声处理30min,放冷,用70%甲醇补足减失的重量,摇匀,离心,取上清液,即得待测产品溶液。(2)参照物溶液的制备:取黄芩苷对照品适量,加甲醇制成每1ml含50μg的溶液,即得。(3)重复6次获取上述溶液的指纹图谱,得到6个指纹图谱,指纹图谱的获取方法为:以浓度为0.2vol%磷酸溶液a、甲醇b、乙腈d为流动相,分别获取所有批次的待测产品溶液的高效液相指纹图谱;色谱条件为:采用welchlp-c18色谱柱,色谱柱长度为250mm,色谱柱内径为4.6mm,填料粒径为5μm;线性梯度洗脱程序为:0~22min内,流动相中a的体积分数为76%,流动相中b的体积分数为1%,流动相中d的体积分数为23%;22~48min内,流动相中a的体积分数为76%~40%,流动相中b的体积分数为1%~50%,流动相中d的体积分数为23%~10%;48~65min内,流动相中a的体积分数为40%~12%,流动相中b的体积分数为50%~33%,流动相中d的体积分数为10%~55%;65~68min内,流动相中a的体积分数为12%,流动相中b的体积分数为33%,流动相中d的体积分数为55%;68~72min内,流动相中a的体积分数为12%~76%,流动相中b的体积分数为33%~1%,流动相中d的体积分数为55%~23%;流动相流速为1.0ml/min;柱温为30℃;进样量为10μl;检测波长为225nm。(4)质量稳定性评价:步骤2得到的6个指纹图谱,结果见图9,图谱的相关数据见表1-1和表1-2;以5号色谱峰(黄芩苷)为参照,计算各共有峰的相对保留时间和相对峰面积,相对保留时间rsd均小于0.23%,相对峰面积rsd均小于0.99%。表明该方法的精密度良好。表1-1精密度实验结果(相对保留时间)共有峰序号123456rsd(%)10.6120.6120.6110.6120.6100.6100.1520.6430.6420.6420.6420.6410.6400.1530.6690.6690.6680.6680.6670.6670.1540.7090.7080.7080.7080.7070.7060.1651.0001.0001.0001.0001.0001.0000.0061.0861.0861.0841.0841.0831.0820.1471.1961.1951.1931.1931.1921.1910.1581.4681.4681.4661.4661.4661.4660.0791.6881.6891.6861.6871.6861.6840.10101.7311.7321.7301.7301.7291.7300.06112.0102.0122.0092.0102.0082.0070.08122.5442.5452.5422.5432.5412.5340.15132.9972.9972.9922.9932.9902.9790.22143.0293.0303.0253.0263.0223.0120.21153.1103.1103.1053.1063.1033.0920.22163.1763.1763.1723.1733.1693.1580.22173.4503.4503.4453.4463.4423.4290.23183.5613.5623.5573.5583.5543.5400.23表1-2精密度实验结果(相对峰面积)2、重复性试验(1)制备同一批次中6份一清颗粒供试品溶液,每份供试品溶液的制备过程如下:取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声处理30min,放冷,用70%甲醇补足减失的重量,摇匀,离心,取上清液,即得6份待测产品溶液。(2)参照物溶液的制备:取黄芩苷对照品适量,加甲醇制成每1ml含50μg的溶液,即得。(3)将上述供试品溶液和参照物溶液注入液相色谱仪,色谱条件同精密度试验项下方法,获取上述6份待测产品溶液的指纹图谱,结果见图10,图谱的相关数据见表2-1和表2-2;以5号色谱峰(黄芩苷)为参照,计算各共有峰的相对保留时间和相对峰面积,相对保留时间rsd均小于0.32%,相对峰面积rsd均小于1.29%。表明该方法重复性良好。表2-1重复性实验结果(相对保留时间)表2-2重复性实验结果(相对峰面积)共有峰序号123456rsd(%)10.0240.0240.0240.0250.0240.0250.8820.0480.0480.0480.0480.0480.0490.8130.0270.0270.0270.0270.0270.0270.7440.0970.0980.0970.0970.0980.0980.4551.0001.0001.0001.0001.0001.0000.0060.0710.0720.0710.0720.0720.0720.7370.3100.3170.3160.3160.3160.3170.9180.0460.0460.0450.0450.0460.0450.9490.0330.0340.0330.0330.0330.0330.41100.1030.1030.1030.1030.1030.1030.21110.1950.1940.1960.1960.1940.1940.49120.1730.1730.1730.1730.1720.1720.41130.0090.0090.0090.0090.0090.0091.25140.0300.0300.0300.0300.0300.0300.29150.0140.0140.0140.0150.0140.0140.38160.0480.0480.0480.0490.0480.0480.36170.0110.0110.0110.0110.0110.0110.49180.0090.0090.0090.0090.0090.0091.293、稳定性试验(1)取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声处理30min,放冷,用70%甲醇补足减失的重量,摇匀,离心,取上清液,即得待测产品溶液。(2)参照物溶液的制备:取黄芩苷对照品适量,加甲醇制成每1ml含50μg的溶液,即得。(3)将该份待测产品溶液,分别在室温放置0h、3h、7h、13h、17h、24h、36h、48h、72h后进样分析,色谱分析方法同精密度试验项下方法,得到9份指纹图谱,结果见图11,相关数据见表3-1和3-2。结果以5号色谱峰(黄芩苷)为参照,计算各共有峰的相对保留时间和相对峰面积,相对保留时间rsd均小于0.47%,相对峰面积rsd均小于2.00%。表明通过该方法提取的一清颗粒供试品溶液在室温72h内样品稳定性良好。表3-1稳定性实验结果(相对保留时间)共有峰序号0h3h7h13h17h24h36h48h72hrsd(%)10.6120.6110.6100.6080.6090.6060.6050.6040.6050.4520.6430.6420.6400.6390.6390.6370.6360.6350.6360.4530.6690.6680.6670.6650.6650.6630.6620.6610.6620.4540.7090.7080.7060.7050.7050.7020.7020.7000.7020.4151.0001.0001.0001.0001.0001.0001.0001.0001.0000.0061.0861.0841.0821.0801.0791.0771.0731.0731.0730.4771.1961.1931.1911.1911.1881.1851.1831.1811.1830.4381.4681.4661.4661.4661.4681.4651.4651.4661.4670.0691.6881.6861.6841.6871.6881.6841.6821.6851.6880.12101.7311.7301.7301.7311.7321.7301.7271.7311.7320.08112.0102.0092.0072.0092.0132.0072.0042.0102.0140.15122.5442.5422.5342.5382.5492.5322.5302.5382.5480.26132.9972.9922.9792.9833.0022.9772.9752.9852.9960.33143.0293.0253.0123.0163.0343.0093.0073.0183.0300.33153.1103.1053.0923.0953.1153.0893.0873.0983.1100.33163.1763.1723.1583.1623.1813.1553.1533.1643.1760.33173.4503.4453.4293.4333.4563.4263.4243.4353.4490.34183.5613.5573.5403.5443.5683.5373.5353.5483.5620.34表3-2稳定性实验结果(相对峰面积)应用实施例1:浙江凯润药业股份有限公司自制的一清颗粒稳定性评价(1)取20批次一清颗粒样品(浙江凯润药业股份有限公司),按照以下方法制备20批次的待测产品溶液:取一清颗粒样品,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入加70%甲醇50ml,称重,超声处理30min,放冷,用70%甲醇补足减失的重量,摇匀,离心,取上清液,即得待测产品溶液。(2)参照物溶液的制备:取黄芩苷对照品适量,加甲醇制成每1ml含50μg的溶液,即得。(3)以浓度为0.2vol%磷酸溶液a、甲醇b、乙腈d为流动相,分别获取所有批次的待测产品溶液的高效液相指纹图谱;色谱条件为:采用welchlp-c18色谱柱,色谱柱长度为250mm,色谱柱内径为4.6mm,填料粒径为5μm;线性梯度洗脱程序为:0~22min内,流动相中a的体积分数为76%,流动相中b的体积分数为1%,流动相中d的体积分数为23%;22~48min内,流动相中a的体积分数为76%~40%,流动相中b的体积分数为1%~50%,流动相中d的体积分数为23%~10%;48~65min内,流动相中a的体积分数为40%~12%,流动相中b的体积分数为50%~33%,流动相中d的体积分数为10%~55%;65~68min内,流动相中a的体积分数为12%,流动相中b的体积分数为33%,流动相中d的体积分数为55%;68~72min内,流动相中a的体积分数为12%~76%,流动相中b的体积分数为33%~1%,流动相中d的体积分数为55%~23%;流动相流速为1.0ml/min;柱温为30℃;进样量为10μl;检测波长为225nm。(4)质量稳定性评价:将步骤(3)得到的20批次待测产品溶液的指纹图谱(如图12),导入“中药色谱指纹图谱相似度评价系统(2012.130723版本)”,经多点校正后进行mark峰匹配,用平均数法(时间窗为0.1min)生成对照指纹图谱(如图13),并计算相似度。指纹图谱包括18个共有峰,经与对照品比对,5号峰为黄芩苷,6号峰为盐酸巴马汀,7号峰为盐酸小檗碱,14号峰为汉黄芩素,16号峰为大黄酸,17号峰为大黄素,18号峰为大黄酚。各批次一清颗粒指纹图谱相似度分别为:0.981、0.983、0.984、0.997、0.998、0.996、0.992、0.999、0.998、0.995、0.994、0.975、0.970、0.999、0.997、0.999、0.999、0.999、0.999、0.937,相似度均大于0.90,结果表明:浙江凯润药业股份有限公司生产的一清颗粒各批次间质量均一、稳定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1