一种青霉素V酸含量的检测方法与流程
本发明涉及药物检测
技术领域:
,尤其涉及一种青霉素v酸含量的检测方法。
背景技术:
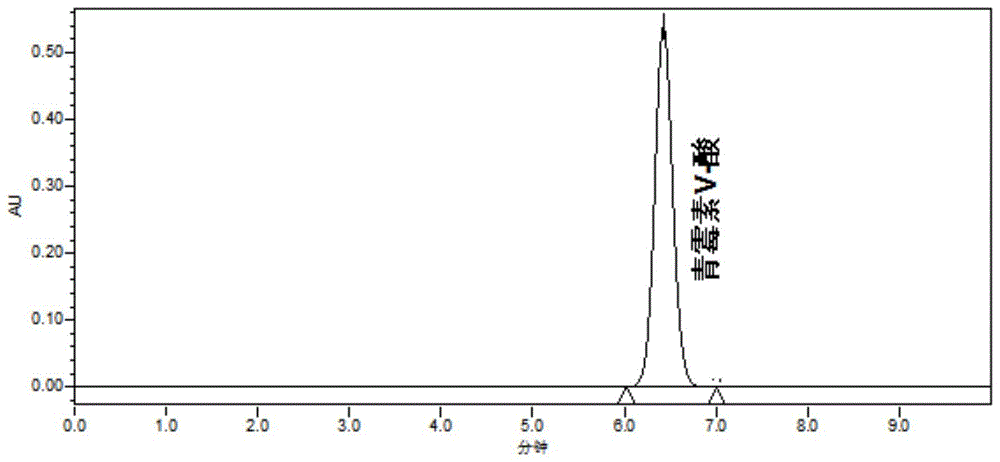
:青霉素v钾用于治疗对青霉素g敏感的轻度、中度、严重感染,适用于外科手术及预防风湿热、霍乱的复发及预防细菌性心内膜炎,敏感菌引起的耳、鼻和咽部感染、呼吸道感染、皮肤感染等。青霉素-v钾具有毒性很小,口服后吸收迅速,食物对药物吸收的影响不显著,抗菌作用强的优点。青霉素v钾的萃取和精制是利用青霉素v-酸在不同的ph值条件下,会以不同的化学状态(青霉素v的游离酸和青霉素v的盐类)存在,利用不同化学状态的青霉素v化合物在水中和有机溶剂中溶解度的差别经过萃取、转移、分离过程,达到浓缩和提纯青霉素v钾的目的。青霉素v酸晶是青霉素v钾生产过程中非常重要的一个中间产品。目前,青霉素v酸晶主要是通过以下工艺制备得到的:向青霉素v的水溶液中,加入稀酸,随着ph的降低,青霉素v的游离酸形成结晶析出,将悬浮液过滤得到青霉素v酸晶,再与碳酸钾反应后过滤、结晶得到成品青霉素v钾。青霉素v酸晶产品中青霉素v酸含量的高低直接关系到青霉素v钾产品质量的好坏。因此,研发一种可以快速准确地检测青霉素v酸含量的方法,对于青霉素v酸晶生产工艺的控制以及青霉素v钾质量的提高具有十分重要的意义。技术实现要素:针对现有技术中存在的上述技术问题,本发明提供一种青霉素v酸含量的检测方法。为解决上述技术问题,本发明提供的技术方案是:一种青霉素v酸含量的检测方法,所述检测方法为高效液相色谱法,其中色谱条件为:色谱柱:十八烷基键合硅胶柱;流动相:甲醇-水-三氟乙酸;检测波长:268nm;洗脱方式为等度洗脱。青霉素v酸晶是生产过程中的中间体产品,产品中杂质成分多,基质干扰大,检测青霉素v酸晶产品中青霉素v酸的含量存在需要样品前处理以及提高检测分离度的难题。本发明提供的青霉素v酸含量的检测方法,采用十八烷基键合硅胶柱色谱柱,甲醇-水-三氟乙酸作为流动相,通过hplc法进行检测,能够快速、准确地对青霉素v酸进行定量检测,出峰时间快,仅8-9min即可完成检测,准确度高,大大提高了生产效率;且青霉素v酸可与其他杂质峰达到很好的分离效果。本发明能够用更为简便的方法实现对青霉素v酸晶体中青霉素v酸的定量检测,且方法简单,操作简便,分离效果好,灵敏度高,检测限低,为提高和更好地控制青霉素v钾产品质量提供了可靠保障,具有广阔的应用前景。中国药典和国外药典已经规定了青霉素v钾的高效液相色谱检测的方法,其中采用十八烷基硅烷键合硅胶为色谱柱;以ph3.5的磷酸盐缓冲液-甲醇-水(10:30:60)为流动相a,以ph3.5的磷酸盐缓冲液-甲醇-水(10:55:35)为流动相b,以流动相a-流动相b(60:40)等度洗脱。在高效液相色谱法分析时,发明人发现,虽然青霉素v钾与青霉素v酸结构相似,但是,采用中国药典和国外药典的方法无法实现对青霉素v酸的定量检测。本发明选择甲醇-水-三氟乙酸作为流动相,通过进一步控制检测条件,从而实现了对青霉素v酸的快速、准确地定量检测。本发明中所述青霉素v酸指的是青霉素v水溶液酸化得到的青霉素v酸。本发明中所用甲醇和三氟乙酸均为市售分析纯的甲醇和三氟乙酸。优选的,所述流动相中甲醇与水的体积比为600:400-800:200,以甲醇和水的总体积为100%计,三氟乙酸的体积百分比为0.3-0.4%。本发明优选的流动相不但可保证青霉素v酸良好的峰形,缩短出峰时间,将其作为溶解青霉素v酸的溶剂,还能有效去除青霉素v酸晶中蛋白和杂质的干扰,提高样品溶液的稳定性,提高检测的分离度和准确度。优选的,所述流动相中甲醇、水和三氟乙酸的体积比为700:300:3.5。优选的流动相比例,可使在不产生基线干扰的前提下,能够更好地分离青霉素v酸和杂质,并且有效改善峰形,使检测结果的准确度及精密度更高。优选的,柱温为30-40℃,进样体积为20μl。优选的,流速为0.8-1.2ml/min。更优选的,流速为1.0ml/min,柱温为40℃。优选的检测条件可使青霉素v酸与杂质之间达到更高的分离度,进而提高检测的准确度,并缩短青霉素v酸的出峰时间,提高检测效率。优选的,高效液相检测的检测器为紫外检测器或dad检测器。优选的检测器可提高分析灵敏度。优选的,所述色谱柱为welchultimatexb-c184.6×250,5μm。优选的色谱柱规格可以使青霉素素v酸的峰形、青霉素v酸与杂质的分离度及检测的灵敏度俱佳,且基线干扰较小,从而有利于青霉素v酸与杂质有效分离,且结果准确可靠,重复性好。优选的,供试品溶液的浓度为1mg/ml。优选的供试品浓度有利于使青霉素v酸及杂质的峰形更好,柱效高,积分更准确,从而有利于对供试品中青霉素v酸的含量进行更准确的计算。附图说明图1为本发明实施例1中浓度为1mg/ml的标准对照品溶液的色谱图;图2为本发明实施例1中浓度为1mg/ml的供试品溶液的色谱图;具体实施方式为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。本发明实施例提供一种青霉素v酸含量的检测方法,步骤如下:步骤1、将青霉素v酸对照品溶于流动相中,按比例稀释,分别相当于每1ml中含0.20mg、0.40mg、0.60g、0.80g、1.0mg、1.20mg的标准溶液;步骤2、采用高效液相色谱仪分别对不同浓度的标准溶液进行液相色谱分析,记录分析得到的峰面积,利用外标法绘制青霉素v酸含量-峰面积的标准曲线,得出标准曲线回归方程;步骤3、将烘干后的青霉素v酸晶样品50mg,用流动相稀释至50ml,利用液相色谱仪进行液相色谱分析,采用与标准溶液完全相同的测定条件,并记录峰面积,根据步骤2的标准曲线回归方程计算得出所述青霉素v酸的含量。检测方法的参数为:色谱柱:十八烷基键合硅胶柱;流动相:甲醇-水-三氟乙酸;其中,所述流动相中甲醇与水的体积比为600:400-800:200,以甲醇和水的总体积为100%计,三氟乙酸的体积百分比为0.3-0.4%;检测波长:268nm;柱温为30-40℃;进样体积为20μl;流速为0.8-1.2ml/min;洗脱方式为等度洗脱。实施例11.材料与方法:仪器:waterse2695高效液相色谱仪,紫外检测器,电子天平。试剂:分析纯甲醇、分析纯三氟乙酸、水、自制青霉素v酸对照品(纯度98.9%,由河北华北制药华恒药业有限公司提供)。空白溶剂:纯化水2.色谱条件色谱柱:welchultimatexb-c184.6×250,5μm;流动相:体积比为700:300:3.5的甲醇-水-三氟乙酸;检测波长:268nm;柱温为40℃;进样体积为20μl;流速为1.0ml/min;洗脱方式为等度洗脱。3.实验步骤:(1)配制流动相,脱气,过滤;(2)调节高效液相色谱仪,平衡色谱柱;(3)对照品溶液的配制,称取青霉素v酸对照品500mg于50ml容量瓶中,用流动相溶解并稀释至刻度,得青霉素v酸标准储备液10mg/ml;分别吸取青霉素v酸标准储备液1.00ml、2.00ml、3.00ml、4.00ml、5.00ml、6.00ml于50ml容量瓶中,用流动相稀释并定容,配制成浓度分别为0.20mg/ml、0.40mg/ml、0.60mg/ml、0.80mg/ml、1.00mg/ml、1.20mg/ml的标准对照品溶液系列,过滤,备用;(4)供试品溶液的配制:经青霉素v酸晶样品50mg加流动相稀释至50.0ml,取10ml于高速离心机离心5-10min,上清液过0.45μm有机微孔滤膜过滤,备用;(5)将标准对照品溶液系列和供试品溶液注入高效液相色谱仪,记录色谱图及峰面积,如图1-2所示,青霉素v酸对照品的出峰时间为6.356min,供试品溶液的出峰时间为6.378min。系列标准对照品溶液的峰面积如表1所示,利用外标法得出标准曲线回归方程y=1.004×107x-5.16×105,r2=0.9999,根据工作曲线计算供试品中青霉素v酸的含量为95.8%。表1为了更好地指导实际生产,在测试供试品时,可将烘干后的青霉素v酸晶供试品样品分为两份,一份用于高效液相含量的测定,一份用于干燥失重的测定,在利用高效液相色谱法测定青霉素v酸的含量后,根据干燥失重测定的水分含量进行折干校正处理,以便更好地指导后续生产。方法学验证:2.1精密度为验证上述实施例1的可行性和准确性,进行精密度试验,试验采用浓度为1.0mg/ml配制的标准对照品溶液,连续进样6次,采用与实施例1完全相同的色谱条件,测定结果表2所示。表2精密度试验结果序号青霉素v酸峰面积195620872961367239518437495280635959256269522865平均值9556281rsd0.42%试验结果表明,峰面积的rsd小于1.0%,因此仪器稳定性良好。2.2线性范围精密称取青霉素v酸对照品适量,分别配制从定量限为起点的一些列浓度的溶液。精密量取上述对照品溶液各20μl分别注入高效液相色谱仪,记录色谱图,测定峰面积,以峰面积a为纵坐标、浓度c为横坐标作线性回归,y=1.0×107x-5.18×105,r2=0.9999。结果表明,青霉素v酸在浓度为0.1~2.0mg/ml范围内r2=0.9999,浓度和峰面积之间线性关系良好。2.3准确度回收率80%溶液:准确称取青霉素v酸对照品40mg,置于50ml容量瓶中,加流动相超声溶解并稀释至刻度,制成每1ml中含青霉素v酸供试品0.8mg的溶液。回收率100%溶液:准确称取青霉素v酸对照品50mg,置于50ml容量瓶中,加流动相超声溶解并稀释至刻度,制成每1ml中含青霉素v酸供试品1.0mg的溶液。回收率120%溶液:准确称取青霉素v酸对照品60mg,置于50ml容量瓶中,加流动相超声溶解并稀释至刻度,制成每1ml中含青霉素v酸供试品1.2mg的溶液。回收率80%溶液、回收率100%溶液和回收率120%溶液各平行配制3份。精密量取上述溶液各20μl,注入液相色谱仪,记录色谱图,结果见下表3。表3回收率试验结果以上结果表明,各杂质在各浓度下的回收率均在99.35-100.30%范围内,rsd值均小于1%,说明本法准确度较好。2.5稳定性试验取供试品溶液,分别于0、1、2、3、4、5h进样,测定青霉素v酸的峰面积,结果见下表4,rsd为1.1%。结果表明,供试品溶液在5h内稳定。表4时间0h1h2h3h4h5h平均rsd%峰面积95620879619012941603795720529388692942102894964861.1实施例2色谱条件色谱柱:welchultimatexb-c184.6×250,5μm;流动相:体积比为800:200:3的甲醇-水-三氟乙酸;检测波长:268nm;柱温为35℃;进样体积为20μl;流速为0.8ml/min;洗脱方式为等度洗脱。实验步骤:(1)配制流动相,脱气,过滤;(2)调节高效液相色谱仪,平衡色谱柱;(3)对照品溶液的配制,称取青霉素v酸对照品500mg于50ml容量瓶中,用流动相溶解并稀释至刻度,得青霉素v酸标准储备液;分别量取青霉素v酸标准储备液1.00ml、2.00ml、3.00ml、4.00ml、5.00ml、6.00ml于50ml容量瓶中,用流动相稀释并定容,配制成浓度分别为0.20mg/ml、0.40mg/ml、0.60mg/ml、0.80mg/ml、1.00mg/ml、1.20mg/ml的标准对照品溶液系列,过滤,备用;(4)供试品溶液的配制:准确称取青霉素v酸晶样品50mg加流动相稀释至50.0ml,取10ml于高速离心机离心5-10min,上清液过0.45μm有机微孔滤膜过滤,备用;(5)将标准对照品溶液系列和供试品溶液注入高效液相色谱仪,记录色谱图及峰面积,青霉素v酸对照品的出峰时间为5.278min,供试品溶液的出峰时间为5.274min。系列标准对照品溶液的峰面积如表5所示,利用外标法得出标准曲线回归方程y=7.98×106x-5.45×105,r2=0.9995,根据工作曲线计算供试品中青霉素v酸的含量为95.3%。表5实施例3色谱条件色谱柱:welchultimatexb-c184.6×250,5μm;流动相:体积比为600:400:4的甲醇-水-三氟乙酸;检测波长:268nm;柱温为30℃;进样体积为20μl;流速为1.2ml/min;洗脱方式为等度洗脱。实验步骤:(1)配制流动相,脱气,过滤;(2)调节高效液相色谱仪,平衡色谱柱;(3)对照品溶液的配制,准确称取青霉素v酸对照品500mg于50ml容量瓶中,用流动相溶解并稀释至刻度,得青霉素v酸标准储备液;分别量取青霉素v酸标准储备液1.00ml、2.00ml、3.00ml、4.00ml、5.00ml、6.00ml于50ml容量瓶中,用流动相稀释并定容,配制成浓度分别为0.20mg/ml、0.40mg/ml、0.60mg/ml、0.80mg/ml、1.00mg/ml、1.20mg/ml的标准对照品溶液系列,过滤,备用;(4)供试品溶液的配制:准确称取青霉素v酸晶样品50mg加流动相稀释至50.0ml,取10ml于高速离心机离心5-10min,上清液过0.45μm有机微孔滤膜过滤,备用;(5)将标准对照品溶液系列和供试品溶液注入高效液相色谱仪,记录色谱图及峰面积,青霉素v酸对照品的出峰时间为8.241min,供试品溶液的出峰时间为8.243min。系列标准对照品溶液的峰面积如表6所示,利用外标法得出标准曲线回归方程y=1.73×107x-9.70×105,r2=0.9996,根据工作曲线计算供试品中青霉素v酸的含量为96.2%。表6本申请实施例2-3中对照品和供试品溶液的峰形与实施例1基本相同。应用效果选择不同条件合成的3批青霉素v酸晶进行样品检测,采用本发明提供的青霉素v酸含量的检测方法进行检测,色谱条件与实施例1相同。按照实施例1的方法将青霉素v酸晶配制为1mg/ml的供试品溶液,过滤后进样检测,测定含量分别为96.8%、95.2%、96.3%,rsd(n=4)分别为0.6%、1.2%、1.1%。对比例1将本发明实施例1流动相中的水替换为等体积的磷酸盐缓冲溶液,其他色谱条件不变,取1.0mg/ml供试品溶液进行检测,连续进样6次,记录峰面积,rsd为3.7%。以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1