N-(苯基磺酰基)苯甲酰胺化合物的HPLC分析方法与流程
本发明属于药物分析技术领域,具体而言,涉及n-(苯基磺酰基)苯甲酰胺化合物的hplc分析方法。
背景技术:
细胞凋亡是程序性细胞死亡的过程,是组织稳态的必需生物学过程。在哺乳动物中,它已被证明可以调节早期胚胎发育。在生命的晚期,细胞死亡是一种默认机制,通过该机制除去潜在危险细胞,例如携带癌症缺陷的细胞。已知若干种凋亡途径。最重要的凋亡途径之一涉及bcl-2蛋白家族,其是细胞凋亡的线粒体(也称为“内在”)途径的关键调节剂。参见danial和korsmeyer,cell776:205-219(2004)。结构同源结构域bh1、bh2、bh3和bh4是bcl-2家族蛋白的特征。bcl-2蛋白质家族可以进一步分为三个亚族,这取决于每种蛋白质含有多少同源结构域及其生物活性,即它是否具有促凋亡或抗细胞凋亡功能。
bcl-2蛋白的第一亚组含有具有全部四个同源结构域的蛋白质,即bh1、bh2、bh3和bh4。它们的一般作用是抗细胞凋亡,即保护细胞免于开始细胞死亡过程。诸如bcl-2、bcl-w、bcl-xl、mcl-1和bfl-1/al的蛋白质是该第一亚组的成员。属于bcl-2蛋白的第二亚组的蛋白质含有三个同源结构域bh1、bh2和bh3,并具有促凋亡作用。第二个亚组的两个主要代表性蛋白质是bax和bak。bcl-2蛋白的第三个亚组由仅含有bh3结构域的蛋白质组成,该亚组的成员通常被称为“仅bh3蛋白质”。它们对细胞的生物学作用是促细胞凋亡的。bim、bid、bad、bik、noxa、hrk、bmf和puma是第三个蛋白质亚家族的例子。
失调的凋亡途径涉及许多重要疾病的病理学,例如神经变性病症(上调的细胞凋亡),例如阿尔茨海默氏病;和增殖性疾病(下调的细胞凋亡),例如癌症、自身免疫疾病和促血栓形成病症。
下调的细胞凋亡(更具体地,bcl-2蛋白质家族)可能参与癌性恶性肿瘤的发作。研究表明,例如,抗凋亡蛋白bcl-2和bcl-xl在许多癌细胞类型中过表达。参见zhang,naturereviewsdrugdiscovery1:101(2002);kirkin等,biochimicaetbiophysicaacta1644:229-249(2004);和amundson等,cancerresearch60:6101-6110(2000)。这种失调的作用是改变细胞的存活,否则细胞会在正常条件下发生细胞凋亡。与不受调节的增殖相关的这些缺陷的重复被认为是癌症进化的起点。这些发现使得用于靶向癌症的药物发现成为可能的新策略。
wo2018/027097a1公开了用于治疗对bc1-2蛋白抑制有反应的疾病、障碍或病症(例如癌症)的n-(苯基磺酰基)苯甲酰胺及相关化合物,并具体公开了代表性化合物:n-((4-(((1,4-二噁烷-2-基)甲基)氨基)-3-硝基苯基)磺酰基)-2-((1h-吡咯并[2,3-b]吡啶-5-基)氧基)-4-(4-((6-(4-氯苯基)螺[3.5]壬-6-烯-7-基)甲基)哌嗪-1-基)苯甲酰胺(下称式1化合物),其结构式如下:
式i化合物
该化合物在苯胺处含有一个手性中心,含有一对有效对映体。药物一般是通过与靶点之间的识别和匹配发挥药理活性,在许多情况下,手性化合物的一对对映体在生物体内的药理活性、代谢过程、代谢速率及毒性等存在显著的差异。另外在吸收、分布和排泄等方面也存在差异,还有对映体之间也有可能相互转化。从已知药效方面考虑,可能存在四种不同的情况:
1)只有一种对映体具有所要求的药理活性,而另一种对映体没有药理作用,例如:β-受体阻断剂普萘洛尔(propranolol)的两个对映异构体的体外活性相差98倍,非甾体抗炎药萘普生(naproxen)的(s)-构型对映体的活性比其对映体的活性强35倍。又如天然的尼古丁(nicotine)的毒性要比其非天然的对映体的毒性大得多。
2)一对对映体中的两个化合物都有同等的或近乎同等的药理活性;
3)两种对映体具有不同的药理活性;
4)各对映体药理活性相同但不相等。
为了保证式i化合物的研发和生产质量,需要对原料药及其制剂的质量进行控制。因此,研究获得一种式i化合物的异构体检查和含量测定的检测方法,对控制药品生产质量具有重要意义。
技术实现要素:
本发明的目的在于克服现有技术中缺少式i化合物的分离检测技术,而提供n-(苯基磺酰基)苯甲酰胺化合物的hplc分析方法,该方法能够分离异构体,并能够准确测定异构体的纯度。
本发明提供了一种物质x的hplc分析方法;所述的物质x为如式i-a和/或i-b所示化合物;
所述的hplc分析方法包括以下步骤:手性色谱柱中,用流动相对物质x进行洗脱,即可;所述的手性色谱柱为多糖衍生物耐溶剂型手性色谱柱;所述的手性色谱柱的柱温为25-35℃;所述的洗脱为等度洗脱;所述的洗脱的流速为0.5-3ml/min;所述hplc分析方法的检测波长为200-300nm;
所述的流动相包括烷烃类溶剂、醇类溶剂、卤代烃类溶剂和醚类溶剂,
按体积份数计,所述的烷烃类溶剂为600-800份,所述的醇类溶剂为50-350份,所述的卤代烃类溶剂为30-100份,所述的醚类溶剂为50-200份。
在本发明某些实施方案中,所述的烷烃类溶剂可为本领域常规的烷烃类溶剂,又可为c5-c10烷烃类溶剂,例如正己烷。
在本发明某些实施方案中,所述的醇类溶剂可为本领域常规的醇类溶剂,又可为c1-c3醇类溶剂,例如乙醇和/或异丙醇,优选异丙醇。
在本发明某些实施方案中,所述的卤代烃类溶剂可为本领域常规的卤代烃类溶剂,例如二氯甲烷。
在本发明某些实施方案中,所述的醚类溶剂可为本领域常规的醚类溶剂,例如四氢呋喃。
在本发明某些实施方案中,按体积份数计,所述的烷烃类溶剂可为650-750份,例如700份。
在本发明某些实施方案中,按体积份数计,所述的醇类溶剂可为60-300份,例如70份、100份、130份或300份,优选60-110份,例如60-80份或90-110份。
在本发明某些实施方案中,按体积份数计,所述的卤代烃类溶剂可为40-90份,例如50份或80份,优选40-60份或70-90份。
在本发明某些实施方案中,按体积份数计,所述的醚类溶剂可为70-160份,例如80份、120份或150份,优选140-160份。
在本发明某些实施方案中,所述的流动相还可进一步包括羧酸类溶剂。所述的羧酸类溶剂可为醋酸。按体积份数计,所述的羧酸类溶剂可为0.5-2.5份,又可为0.5-1.5份,例如1份。
在本发明某些实施方案中,所述的流动相还可进一步包括有机胺。所述的有机胺可为二乙胺和/或三乙胺,优选二乙胺。按体积份数计,所述的有机胺可为0.1-4份,又可为0.1-2.5份,例如0.2份、0.5份、1份和2份,优选0.1-1.5份,又可为0.1-0.3份。当所述的有机胺为二乙胺和三乙胺时,所述的二乙胺和三乙胺的体积比可为1:0.5-1:1.5,例如1:1。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂和醚类溶剂组成。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂和醚类溶剂组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-300份,所述的卤代烃类溶剂为40-90份,所述的醚类溶剂为70-160份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂和醚类溶剂组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-110份,所述的卤代烃类溶剂为40-90份,所述的醚类溶剂为140-160份;所述的烷烃类溶剂为正己烷,所述的醇类溶剂为乙醇和/或异丙醇,所述的卤代烃类溶剂为二氯甲烷,所述的醚类溶剂为四氢呋喃。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、乙醇100份、二氯甲烷50份、四氢呋喃150份。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、异丙醇70份、二氯甲烷80份、四氢呋喃150份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂和羧酸类溶剂组成;按体积份数计,所述的烷烃类溶剂为600-800份,所述的醇类溶剂为50-350份,所述的卤代烃类溶剂为30-100份,所述的醚类溶剂为50-200份,所述的羧酸类溶剂为0.5-2.5份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂和羧酸类溶剂组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-300份,所述的卤代烃类溶剂为40-90份,所述的醚类溶剂为70-160份,所述的羧酸类溶剂为0.5-1.5份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂和羧酸类溶剂组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-80份,所述的卤代烃类溶剂为70-90份,所述的醚类溶剂为140-160份,所述的羧酸类溶剂为0.5-1.5份;所述的烷烃类溶剂为正己烷,所述的醇类溶剂为乙醇和/或异丙醇,所述的卤代烃类溶剂为二氯甲烷,所述的醚类溶剂为四氢呋喃,所述的羧酸类溶剂为醋酸。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷00份、异丙醇70份、二氯甲烷80份、四氢呋喃150份、醋酸1份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂和有机胺组成;按体积份数计,所述的烷烃类溶剂为600-800份,所述的醇类溶剂为50-350份,所述的卤代烃类溶剂为30-100份,所述的醚类溶剂为50-200份,所述的有机胺为0.1-4份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂和有机胺组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-300份,所述的卤代烃类溶剂为40-90份,所述的醚类溶剂为70-160份,所述的有机胺为0.1-2.5份;所述的烷烃类溶剂为正己烷,所述的醇类溶剂为乙醇和/或异丙醇,所述的卤代烃类溶剂为二氯甲烷,所述的醚类溶剂为四氢呋喃,所述的有机胺为二乙胺和/或三乙胺。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂和有机胺组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为90-110份,所述的卤代烃类溶剂为40-60份,所述的醚类溶剂为140-160份,所述的有机胺为0.1-1.5份;所述的烷烃类溶剂为正己烷,所述的醇类溶剂为异丙醇,所述的卤代烃类溶剂为二氯甲烷,所述的醚类溶剂为四氢呋喃;所述的有机胺为二乙胺。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、乙醇130份、二氯甲烷50份、四氢呋喃120份、二乙胺1份、三乙胺1份。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、异丙醇100份、二氯甲烷50份、四氢呋喃150份、二乙胺1份、三乙胺1份。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、乙醇150份、异丙醇150份、二氯甲烷50份、四氢呋喃80份、二乙胺1份。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、异丙醇100份、二氯甲烷50份、四氢呋喃150份、二乙胺1份。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、乙醇130份、二氯甲烷50份、四氢呋喃120份、二乙胺2份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂、羧酸类溶剂和有机胺组成;按体积份数计,所述的烷烃类溶剂为600-800份,所述的醇类溶剂为50-350份,所述的卤代烃类溶剂为30-100份,所述的醚类溶剂为50-200份,所述的羧酸类溶剂为0.5-2.5份,所述的有机胺为0.1-4份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂、羧酸类溶剂和有机胺组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-300份,所述的卤代烃类溶剂为40-90份,所述的醚类溶剂为70-160份,所述的羧酸类溶剂为0.5-1.5份,所述的有机胺为0.1-2.5份。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂、羧酸类溶剂和有机胺组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-80份,所述的卤代烃类溶剂为70-90份,所述的醚类溶剂为140-160份,所述的羧酸类溶剂为0.5-1.5份,所述的有机胺为0.1-1.5份;所述的烷烃类溶剂为正己烷,所述的醇类溶剂为乙醇和/或异丙醇,所述的卤代烃类溶剂为二氯甲烷,所述的醚类溶剂为四氢呋喃;所述的羧酸类溶剂为醋酸,所述的有机胺为二乙胺和/或三乙胺。
在本发明某些实施方案中,所述的流动相由烷烃类溶剂、醇类溶剂、卤代烃类溶剂、醚类溶剂、羧酸类溶剂和有机胺组成;按体积份数计,所述的烷烃类溶剂为650-750份,所述的醇类溶剂为60-80份,所述的卤代烃类溶剂为70-90份,所述的醚类溶剂为140-160份,所述的羧酸类溶剂为0.5-1.5份,所述的有机胺为0.1-0.3份;所述的烷烃类溶剂为正己烷,所述的醇类溶剂为异丙醇,所述的卤代烃类溶剂为二氯甲烷,所述的醚类溶剂为四氢呋喃;所述的羧酸类溶剂为醋酸,所述的有机胺为二乙胺。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、异丙醇70份、二氯甲烷80份、四氢呋喃150份、醋酸1份、二乙胺0.2份。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、异丙醇70份、二氯甲烷80份、四氢呋喃150份醋酸1份、二乙胺0.5份。
在本发明某些实施方案中,所述的流动相由下述组分组成;按体积份数计,正己烷700份、异丙醇70份、二氯甲烷80份、四氢呋喃150份、醋酸1份、二乙胺1份。
在本发明某些实施方案中,所述的物质x可以以溶液的形式添加到所述的手性色谱柱上。所述的物质x的溶液的制备方法可为方法一或方法二:
方法一:将物质x溶于溶剂,即可;
方法二:提取含物质x的制剂,即可。
所述的提取可通过本领域常规的手段进行提取。
所述的溶液中的溶剂可与所述的流动相相同,或为卤代烃类溶剂和所述的流动相的混合溶剂,所述的卤代烃类溶剂和所述的流动相的体积比可为0.5:1-2:1,例如1:1。所述的卤代烃类溶剂可为二氯甲烷。
在本发明某些实施方案中,所述的手性色谱柱可为多糖衍生物耐溶剂型手性色谱柱(其通过化学键合型结合的方式将多糖衍生物固定在硅胶上),又可为硅胶表面键合有纤维素-三(3,5-二氯苯基氨基甲酸酯)的糖衍生物耐溶剂型手性色谱柱,例如daicelchiralpakic手性色谱柱,又例如daicelchiralpakic(250mm×4.6mm,5.0µm)。
在本发明某些实施方案中,所述的手性色谱柱的柱温可为本领域常规的柱温,又可为25-35℃,例如33℃。
在本发明某些实施方案中,所述的物质x的溶液的进样体积可为本领域常规的进样体积,又可为1-30μl,又可为10-20μl。
在本发明某些实施方案中,所述的洗脱可为等度洗脱。
在本发明某些实施方案中,所述的洗脱的流速可为本领域常规的流速,又可为0.5-3ml/min,又可为1-2ml/min。
在本发明某些实施方案中,所述hplc分析方法的检测波长可为本领域常规的检测波长,又可为200-300nm,又可为260-290nm,例如265nm、282nm或287nm。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:本发明所提供的式i化合物的hplc分析方法具有使用可靠,稳定性较好,数据重现性强的优点,可以很好地将异构体分离,并能够准确测定异构体的纯度。
附图说明
图1是实施例1中(r)-式i化合物定位溶液的hplc谱图。
图2是实施例1中(s)-式i化合物定位溶液的hplc谱图。
图3是实施例1中式i化合物分离度溶液的hplc谱图。
图4是实施例1中供试品溶液i的hplc谱图。
图5是实施例2中(r)-式i化合物定位溶液的hplc谱图。
图6是实施例2中(s)-式i化合物定位溶液的hplc谱图。
图7是实施例2中式i化合物分离度溶液的hplc谱图。
图8是实施例3中(r)-式i化合物定位溶液的hplc谱图。
图9是实施例3中(s)-式i化合物定位溶液的hplc谱图。
图10是实施例3中式i化合物分离度溶液的hplc谱图。
图11是实施例3中供试品溶液的hplc谱图。
图12是实施例4中(r)-式i化合物定位溶液的hplc谱图。
图13是实施例4中(s)-式i化合物定位溶液的hplc谱图。
图14是实施例4中式i化合物分离度溶液的hplc谱图。
图15是实施例5中(r)-式i化合物定位溶液的hplc谱图。
图16是实施例5中(s)-式i化合物定位溶液的hplc谱图。
图17是实施例5中式i化合物分离度溶液的hplc谱图。
图18是实施例6中式i化合物分离度溶液的hplc谱图。
图19是实施例7中式i化合物分离度溶液的hplc谱图。
图20是实施例8中式i化合物分离度溶液的hplc谱图。
图21是实施例9中式i化合物分离度溶液的hplc谱图。
图22是实施例10中式i化合物分离度溶液的hplc谱图。
图23是实施例11中式i化合物分离度溶液的hplc谱图。
图24是对比例1中式i化合物分离度溶液的hplc谱图。
图25是对比例2中式i化合物分离度溶液的hplc谱图。
图26是对比例3中式i化合物分离度溶液的hplc谱图。
图27是实施例1供试品溶液ii的hplc谱图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
下述实施例中,(r)-式i化合物参照专利申请cn109311871a制得。(s)-式i化合物、式i化合物(r和s构型的混合物)参照(r)-式i化合物的制备方法进行制备,换用原料即可。本发明通过本领域常规的手段制得式i化合物的片剂。
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm),购买厂家:江苏明捷科学仪器有限公司;生产厂家:大赛璐(daicel)。
实施例1:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:正己烷/异丙醇/二氯甲烷/四氢呋喃/醋酸/二乙胺=700/70/80/150/1/0.2。
检测波长:282nm。
流速:2.0ml/min。
柱温33℃。
进样体积:20μl。
稀释剂:二氯甲烷与流动相体积配比1:1。
实验步骤
溶液配制:取式i化合物约20mg,精密称定置于10ml量瓶中,加入5ml二氯甲烷,待样品完全溶解后,用流动相稀释至刻度,摇匀,作为供试品溶液i;
取式i化合物的片剂1片研磨成细粉,转移至10ml容量瓶中,加入5ml二氯甲烷,超声5min,用流动相稀释至刻度,经有机滤膜过滤,取滤液作为供试品溶液ii;
称量(s)-式i化合物对照品约20mg,置于100ml量瓶中,加入50ml二氯甲烷,待完全溶解后,用流动相稀释至刻度,摇匀,再从此溶液中精密量取1ml置于100ml量瓶中,加稀释剂至刻度,摇匀,作为(s)-式i化合物定位溶液;
称量(r)-式i化合物对照品约20mg,置于100ml量瓶中,加50ml二氯甲烷溶解后,用流动相稀释至刻度,摇匀,再从此溶液中精密量取1ml置100ml量瓶中,加稀释剂至刻度,摇匀,作为(r)-式i化合物定位溶液;
分别称量(s)-式i化合物对照品约20mg、(r)-式i化合物对照品约1mg置于同一10ml量瓶中,加5ml二氯甲烷溶解后,用流动相稀释至刻度,摇匀,作为分离度溶液。
精密量取(s)-式i化合物定位溶液、(r)-式i化合物定位溶液、分离度溶液、供试品溶液各20μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表1)。
表1系统适用性测试结果
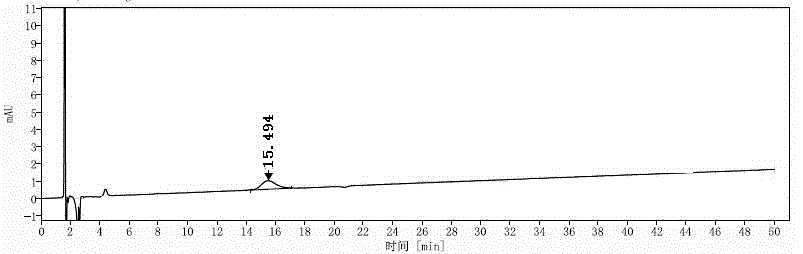
根据表1及图1-4、图27可以看出,(r)-式i化合物与(s)-式i化合物可以得到有效分离,分离度为1.9,峰形良好,峰纯度良好,符合标准。
本发明所提供的分析方法,根据中国药典2015版,完成方法学验证,验证系统适用性、专属性、灵敏度、线性和范围、精密度、准确度、重现性以及溶液稳定性实验。经过这些验证,本发明提供的分析方法使用可靠,稳定性较好。
实施例2:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:二乙胺/三乙胺/四氢呋喃/乙醇/二氯甲烷/正己烷=1/1/120/130/50/700。
检测波长:287nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:20μl。
稀释剂:组成同流动相。
实验步骤
溶液配制:取式i化合物约20mg,精密称定置于50ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,作为供试品溶液;
取(s)-式i化合物对照品约20mg,精密称定置于50ml量瓶中,用稀释剂溶解后稀释至刻度,摇匀,精密量取1ml溶液置20ml量瓶,用稀释剂稀释至刻度,摇匀,再精密量取1ml此溶液置20ml量瓶,用稀释液稀释至刻度,摇匀,作为(s)-式i化合物定位溶液;
取(r)-式i化合物对照品约20mg,精密称定置于50ml量瓶中,用稀释剂溶解后稀释至刻度,摇匀,精密量取1ml溶液置20ml量瓶,用稀释剂稀释至刻度,摇匀,再精密量取1ml此溶液置20ml量瓶,用稀释液稀释至刻度,摇匀,作为(r)-式i化合物定位溶液;
取(s)-式i化合物对照品约20mg,精密称定,置50ml量瓶中,量取(r)-式i化合物定位溶液原液1ml,稀释剂溶解后,稀释至刻度,摇匀,作为分离度溶液。精密量取(s)式i化合物定位溶液、(r)-式i化合物定位溶液、分离度溶液各20μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表2)。
表2系统适用性测试结果
根据表2及图5-7可以看出,(r)-式i化合物与(s)-式i化合物可以分离,分离度为1.0,峰纯度良好。
实施例3:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:四氢呋喃/乙醇/二氯甲烷/正己烷=150/100/50/700。
检测波长:282nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:10μl。
稀释剂:二氯甲烷与流动相体积比为1:1。
实验步骤
溶液配制:取式i化合物约30mg,精密称定置于20ml量瓶中,用10ml二氯甲烷溶解后,用流动相稀释至刻度,摇匀,作为供试品溶液;
取(s)-式i化合物对照品约30mg,精密称定置于20ml量瓶中,用10ml二氯甲烷溶解后,用流动相稀释至刻度,摇匀,精密量取溶液1ml至50ml量瓶中,用稀释剂稀释至刻度,摇匀,再精密量取此溶液1ml置20ml量瓶中,用稀释剂稀释至刻度,摇匀,作为(s)-式i化合物定位溶液;
取(r)-式i化合物对照品约30mg,精密称定置于20ml量瓶中,用10mldcm溶解后,用流动相稀释至刻度,摇匀,精密量取溶液1ml至50ml量瓶中,用稀释剂稀释至刻度,摇匀,再精密量取此溶液1ml置20ml量瓶中,用稀释剂稀释至刻度,摇匀,作为(r)-式i化合物定位溶液;
取(s)-式i化合物约30mg,精密称定置于20ml量瓶中,量取(r)-式i化合物定位溶液原液2ml,加10ml二氯甲烷溶解,用流动相稀释至刻度,摇匀,作为分离度溶液。精密量取(s)-式i化合物定位溶液、(r)-式i化合物定位溶液、分离度溶液、供试品溶液各10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表3)
表3系统适用性测试结果
根据表3及图8-11可以看出,(r)-式i化合物与(s)-式i化合物可以得到有效分离,分离度为1.7,峰纯度良好,符合标准。
实施例4:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:正己烷/异丙醇/二氯甲烷/四氢呋喃=700/70/80/150。
检测波长:282nm。
流速:2.0ml/min。
柱温:33℃。
进样体积:10μl。
稀释剂:二氯甲烷与流动相体积比为1:1。
实验步骤
溶液配制:取式i化合物约20mg,精密称定置于20ml量瓶中,加入10ml二氯甲烷,待样品完全溶解后,用流动相稀释至刻度,摇匀,作为供试品溶液;
取(s)-式i化合物约20mg,精密称定置于20ml量瓶中,加入10ml二氯甲烷,待样品完全溶解后,用流动相稀释至刻度,摇匀,精密量取溶液1ml至50ml量瓶中,用稀释剂稀释至刻度,摇匀,再精密量取此溶液1ml置20ml量瓶中,用稀释剂稀释至刻度,摇匀,作为(s)-式i化合物定位溶液;
取(r)-式i化合物约20mg,精密称定置于20ml量瓶中,加入10ml二氯甲烷,待样品完全溶解后,用流动相稀释至刻度,摇匀,精密量取溶液2ml至10ml量瓶中,用稀释剂稀释至刻度,摇匀,作为(r)-式i化合物定位溶液;
取(s)-式i化合物约20mg,精密称定置于20ml量瓶中,量取(r)-式i化合物定位溶液原液0.5ml,加10ml二氯甲烷溶解,用流动相稀释至刻度,摇匀,作为分离度溶液。精密量取(s)-式i化合物定位溶液、(r)-式i化合物定位溶液、分离度溶液各10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表4)
表4系统适用性测试结果
根据表4及图12-14可以看出,(r)-式i化合物与(s)-式i化合物可以分离,分离度为1.8,峰形良好,峰纯度良好,符合标准。
实施例5:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:正己烷/异丙醇/二氯甲烷/四氢呋喃/醋酸=700/70/80/150/1。
检测波长:282nm。
流速:2.0ml/min。
柱温33℃。
进样体积:20μl。
稀释剂:二氯甲烷与流动相体积比为1:1。
实验步骤
溶液配制:取式i化合物约30mg,精密称定置于20ml量瓶中,用10ml二氯甲烷溶解后,用流动相稀释至刻度,摇匀,作为供试品溶液;
取(s)-式i化合物对照品约30mg,精密称定置于20ml量瓶中,用10ml二氯甲烷溶解后,用流动相稀释至刻度,摇匀,精密量取溶液1ml至50ml量瓶中,用稀释剂稀释至刻度,摇匀,再精密量取此溶液1ml置20ml量瓶中,用稀释剂稀释至刻度,摇匀,作为(s)-式i化合物定位溶液;
取(r)-式i化合物对照品约30mg,精密称定置于20ml量瓶中,用10ml二氯甲烷溶解后,用流动相稀释,摇匀,精密量取溶液1ml至50ml量瓶中,用稀释剂稀释至刻度,摇匀,再精密量取此溶液1ml置20ml量瓶中,用稀释剂稀释至刻度,摇匀,作为(r)-式i化合物定位溶液;
取(s)-式i化合物约30mg,精密称定置于20ml量瓶中,量取(r)-式i化合物定位溶液原液2ml,加10ml二氯甲烷溶解,用流动相稀释至刻度,摇匀,作为分离度溶液。精密量取(s)-式i化合物定位溶液、(r)-式i化合物定位溶液、分离度溶液各10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表5)。
表5系统适用性测试结果
根据表5及图15-17可以看出,(r)-式i化合物与(s)-式i化合物可以得到有效分离,分离度为2.1,峰形良好,峰纯度良好,符合标准。
实施例6:式i手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:正己烷/异丙醇/二氯甲烷/四氢呋喃/醋酸/二乙胺=700/70/80/150/1/1.0。
检测波长:282nm。
流速:2.0ml/min。
柱温:33℃。
进样体积:20μl。
稀释剂:二氯甲烷与流动相体积配比1:1。
实验步骤
溶液配制:取式i化合物约20mg,精密称定置于10ml量瓶中,加入5mldcm,待样品完全溶解后,用稀释剂稀释至刻度,摇匀,作为供试品溶液;
分别称量(s)-式i化合物对照品约20mg、(r)-式i化合物对照品约1mg置同一10ml量瓶中,加5mldcm溶解后,用流动相稀释至刻度,摇匀,作为分离度溶液。精密量取分离度溶液20μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表6)。
表6系统适用性测试结果
根据表6及图18可以看出,(r)-式i化合物与(s)-式i化合物可以得到有效分离,分离度为1.6,峰形良好,峰纯度良好,符合标准。
实施例7:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:正己烷/异丙醇/二氯甲烷/四氢呋喃/醋酸/二乙胺=700/70/80/150/1/0.5。
检测波长:282nm。
流速:2.0ml/min。
柱温:33℃。
进样体积:20μl。
稀释剂:二氯甲烷与流动相体积配比1:1。
实验步骤
溶液配制:取式i化合物约20mg,精密称定置于10ml量瓶中,加入5ml二氯甲烷,待样品完全溶解后,用流动相稀释至刻度,摇匀,作为供试品溶液;
分别称量(s)-式i化合物对照品约20mg、(r)-式i化合物对照品约1mg置同一10ml量瓶中,加5ml二氯甲烷溶解后,用流动相稀释至刻度,摇匀,作为分离度溶液。精密量取分离度溶液20μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表7)。
表7系统适用性测试结果
根据表7及图19可以看出,(r)-式i化合物与(s)-式i化合物可以得到有效分离,分离度为1.8,峰形良好,峰纯度良好,符合标准。
实施例8:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:四氢呋喃/异丙醇/二氯甲烷/正己烷/二乙胺/三乙胺=150/100/50/700/1/1。
检测波长:265nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:10μl。
稀释剂:组成同流动性。
实验步骤
溶液配制:分别取(r)-式i化合物对照品约2mg和(s)-式i化合物对照品约20mg,精密称定置于20ml量瓶中,摇匀,作为分离度溶液。
精密量取分离度溶液10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表8)
表8系统适用性测试结果
根据表8及图20可以看出,(r)-式i化合物与(s)-式i化合物可以分离,分离度为1.1。
实施例9:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:四氢呋喃/乙醇/异丙醇/二氯甲烷/正己烷/二乙胺=80/150/150/50/700/1。
检测波长:265nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:10μl。
稀释剂:组成同流动性。
实验步骤
溶液配制:分别取(r)-式i化合物对照品和(s)-式i化合物对照品约20mg,精密称定置于20ml量瓶中,用稀释剂溶解后稀释至刻度,摇匀,作为分离度溶液。
精密量取分离度溶液10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表9)
表9系统适用性测试结果
根据表9及图21可以看出,(r)-式i化合物与(s)-式i化合物可以分离,分离度为1.3。
实施例10:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:四氢呋喃/异丙醇/二氯甲烷/正己烷/二乙胺=150/100/50/700/1。
检测波长:265nm。
流速:1.0ml/min。
柱温33℃。
进样体积:10μl。
稀释剂:组成同流动性。
实验步骤
溶液配制:分别取(r)-式i化合物对照品和(s)-式i化合物对照品约20mg,精密称定置于20ml量瓶中,摇匀,作为分离度溶液。
精密量取分离度溶液10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表10)
表10系统适用性测试结果
根据表10及图22可以看出,(r)-式i化合物与(s)-式i化合物可以很好的分离,分离度为2.0。
实施例11:式i手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:四氢呋喃/乙醇/二氯甲烷/正己烷/二乙胺=120/130/50/700/2。
检测波长:265nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:10μl。
稀释剂:组成同流动性。
实验步骤
溶液配制:分别取(r)-式i化合物对照品和(s)-式i化合物对照品约20mg,精密称定置于20ml量瓶中,用稀释剂溶解后稀释至刻度,摇匀,作为分离度溶液。
精密量取分离度溶液10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表11)
表11系统适用性测试结果
根据表11及图23可以看出,(r)-式i化合物与(s)-式i化合物可以分离,分离度为1.5。
对比例1:式i化合物手性纯度的测定
流动相体积配比:乙醇/正己烷/二乙胺=300/700/1。
检测波长:265nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:10μl。
稀释剂:组成同流动相。
实验步骤
溶液配制:分别取(r)-式i化合物对照品和(s)-式i化合物对照品约20mg,精密称定置于20ml量瓶中,用稀释剂溶解后稀释至刻度,摇匀,作为分离度溶液。
精密量取分离度溶液10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算。
根据图24可以看出,(r)-式i化合物和(s)-式i化合物该条件下120min皆未出峰,该条件不合适。
对比例2:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:正己烷/乙醇/二氯甲烷/二乙胺=600/200/200/1。
检测波长:265nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:20μl。
稀释剂:组成同流动相。
实验步骤
溶液配制:分别取(r)-式i化合物对照品和(s)-式i化合物对照品约20mg,精密称定置于50ml量瓶中,摇匀,作为分离度溶液。
精密量取分离度溶液20μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表12)
表12系统适用性测试结果
n/a表示已检测,由于分离度差而无法计算获得。
根据表12及图25可以看出,(r)-式i化合物与(s)-式i化合物分离较差。
对比例3:式i化合物手性纯度的测定
高效液相色谱条件:
色谱柱:daicelchiralpakic(250mm×4.6mm,5.0µm)。
流动相体积配比:四氢呋喃/乙醇/正己烷/二乙胺=80/250/650/1。
检测波长:265nm。
流速:1.0ml/min。
柱温:33℃。
进样体积:10μl。
稀释剂:组成同流动相。
实验步骤
溶液配制:分别取(r)-式i化合物对照品和(s)-式i化合物对照品约20mg,精密称定置于20ml量瓶中,摇匀,作为分离度溶液。
精密量取分离度溶液10μl注入液相色谱仪,记录液相色谱图,按照面积归一化法计算(参见表13)
表13系统适用性测试结果
n/a表示已检测,由于分离度差而无法计算获得。
根据表13及图26可以看出,(r)-式i化合物与(s)-式i化合物分离度较差。
- 还没有人留言评论。精彩留言会获得点赞!