一种酿造酱油氨基酸态氮含量是否符合一定级别的快速检测方法与流程
本发明涉及酿造酱油的质量,尤其涉及一种酿造酱油氨基酸态氮含量是否符合一定级别的快速检测方法。
背景技术:
酿造酱油是以大豆、小麦或麸皮为原料,经一定的工艺和微生物发酵制成的具有特殊色、香、味的液体调味品,它的色泽为红褐色或棕褐色,酱香较浓,味鲜咸适口,营养丰富,是大众日用必备调料。
国家对酿造酱油产品的质量已经颁布了相关标准,在gb18186-2000《酿造酱油》的标准中,规定无论是高盐稀态发酵酱油还是低盐固态发酵酱油,用氨基酸态氮(以氮计)含量高低作为指标,分为特级,一级,二级,三级等四个等级。酿造酱油中,氨基酸态氮含量≥0.80g/100ml为特级;≥0.70g/100ml为一级;≥0.60g/100ml为二级(低盐固态发酵酱油)、≥0.55g/100ml为二级(高盐稀态发酵酱油);≥0.40g/100ml为三级,低于三级的为不合格产品。
国家食品安全标准gb5009.235-2016《食品中氨基酸态氮含量的测定》的第一法为酸度计法,该法用naoh标准溶液将酱油样品处理为碱性(ph=8.2),除去样品中的可滴定的酸性物质,然后加入甲醛,将氨基酸羧基的酸性等物质量的显示出来后,用naoh标准溶液滴定至终点,同时测定空白,根据空白和样品所消耗的naoh的量,代入相应的公式计算结果,根据计算结果判断酿造酱油的质量和相应级别。国标法法存在以下不足:
(1)用酸度计测定时,选用naoh标准溶液滴定不合适,因为酱油含有大量的食盐(氯化钠),而且,随着滴定的进行,由于氢氧化钠标准溶液的加入,又不断增加了大量的钠离子,所用ph玻璃电极会产生显著的“钠差”,使结果明显偏低,引入误差;国标法没有考虑“钠差”对测定结果的影响。
(2)甲醛试剂在运输和放置过程中,常常被空气氧化而含有甲酸,必须处理成中性甲醛,排除可能存在的甲酸的干扰,提高测定准确度,国标法没有考虑此因素的影响。
(3)gb5009.235-2016第一法,加入甲醛转化氨基酸态氮前,试液的ph滴定为8.2,除去酱油中存在的酸性物质,加入甲醛后用naoh标准溶液滴定至ph9.2为终点,违背了分析化学定量分析中的“等物质的量转化和反应原则”,实验设计缺乏科学依据支撑,也没有道理。
(4)所用酸度计需要用合适的ph标准溶液进行校正,玻璃电极需要在蒸馏水中浸泡24h以上方可投入使用;影响测定速度和工作效率。
(5)酸度计工作必须用电源,对无电源场合,应用受到限制,无法工作。
(6)试液温度变化会影响测定结果,需随时对酸度计进行温度校正或通过仪器的相关插口安装温度补偿传感器,以排除温度的干扰,给工作带来诸多不便,也会影响测定的准确度。
(7)国标法所用试液、试剂量较大,投入成本相对较高;
(8)操作相对繁杂,结果判断需代入公式进行计算,结果判断不直观。
(9)从取样、样品处理、滴定到获得计算结果,通常需要2h以上,工作效率较低。
(10)滴定过程中,ph玻璃电极的膜电位达平衡需要一定时间,会出现“终点滞后现象”,容易滴过,引入误差,影响测定结果。
技术实现要素:
为解决上述问题,本发明提供一种酿造酱油氨基酸态氮含量是否符合一定级别的快速检测方法,无需用电源和酸度计等仪器滴定,不存在电极的“钠差”和响应“滞后”现象,操作简单,便捷,无需进行计算,结果判断直观、准确;取样后数分钟内即可获得甄别结果。该法应用范围广,实用性强,经与国标法对照测定,结果完全吻合一致。
本发明的目的是以下述方式实现的:一种酿造酱油氨基酸态氮含量是否符合一定级别的快速检测方法,包括以下步骤,(1)取酿造酱油样品a,并进行脱色,脱色后为脱色后的酱油样品b;(2)取含有v2ml酱油样品a的脱色后的酱油样品b,并向其滴加指示剂,此时溶液显示为指示剂的酸色,然后用胶头滴管滴加0.050mol/l的naoh至溶液颜色显示为指示剂的酸色和碱色的混合色,且半分钟不褪色;然后加入中性甲醛,把所取酱油样品b中的氨基酸态氮完全转化为酸性物质(-cooh),此时溶液为指示剂的酸色;然后加入v3ml的一元强碱溶液,轻轻摇晃后观察溶液颜色,此时,溶液显示为指示剂的酸色或溶液酸碱“等量点”时显示指示剂的酸色和碱色的混合色,且半分钟不褪色则该样品是符合一定级别的合格酿造酱油,若溶液显示为指示剂的碱色且半分钟不褪色则为不符合一定级别的不合格酿造酱油;一元强碱溶液的浓度与酱油样品a所在等级的最低浓度相等,且v2=v3。
一元强碱溶液为氢氧化钠溶液、氢氧化钾溶液或氢氧化锂溶液。
酱油样品a为酱油原样,酱油原样为市场售卖或工厂生产的酱油产品;酱油原样为李锦记味极鲜、厨邦海鲜酱油/生抽、海天酱油老抽王、李锦记精选生抽、李锦记锦珍老抽、加加草菇老抽、加加金标生抽、鲁花自然鲜酱香酱油。
指示剂为紫薯色素。
紫薯色素的制备方法为:取紫薯,并研磨成糊状,按料液比1:5加入蒸馏水,移入100ml比色管中,用超声波辅助提取,超声波功率为500w,提取温度为55℃,时间为15-20min,然后过滤到得到紫薯色素;所用蒸馏水的ph为6.8。
所述步骤(1)中酱油进行脱色为:用移液枪移取1.00ml酱油原样样品于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,得到的过滤后的澄清液为脱色后的酱油样品b。
所述步骤(2)中,用移液枪取1.00ml酱油样品b于10ml比色管中,滴加2至3滴天然紫薯色素,此时溶液为红色或红橙色,用胶头滴管逐滴滴加0.050mol/lnaoh标准溶液至颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色或红橙色。
紫薯色素的酸色显示为红色或红橙色,紫薯色素的碱色显示为灰蓝色、绿土色或黄棕色,被鉴别溶液酸碱“等量点”时显示的是指示剂的红橙色与灰蓝色的混合色,为橙灰色。
各级别产品溶液中余剩的酸性物质的量,红色大于红橙色,红橙色大于橙灰色;溶液中余剩的一元强碱量,黄棕色大于绿土色,绿土色大于灰蓝色。
有益效果:相对于现有技术,本发明提供的酱油氨基酸态氮含量是否合格的快速检测的方法,根据gb18186-2000,《酿造酱油》标准中氨基酸态氮的分级参数指标,依据“等物质量的反应原则”,科学设定了鉴别各级酱油产品所需的naoh浓度和量,优选了脱色剂和天然呈色指示剂。该法无需用电源和酸度计等仪器滴定,不存在电极的“钠差”和响应“滞后”现象,所需试液和标准溶液用量降低10倍左右。本发明提供的一种酿造酱油氨基酸态氮含量是否符合一定级别的快速检测方法,操作简单,便捷,无需进行计算,结果判断直观、准确;取样后数分钟内即可获得甄别结果。该法应用范围广,实用性强,经与国标法对照测定,结果完全吻合一致。
附图说明
图1是紫薯色素在不同ph溶液中的颜色变化,自左至右:ph分别为2.00、3.00、4.00、5.00、6.00、7.00、8.00、9.00、10.00、11.00、12.00和13.00。颜色由红色(ph2、3)、红橙色(ph4、5、6、7)、橙灰色(ph8)、灰蓝色(ph9)、绿土色(ph10、11)至黄棕色(ph12、13)。
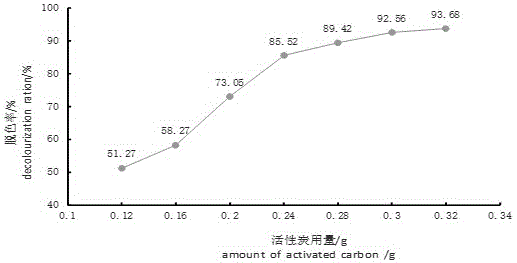
图2是活性炭用量对酱油脱色的影响。
图3是时间对酱油脱色的影响。
具体实施方式
一种酿造酱油氨基酸态氮含量是否符合一定级别的快速检测方法,包括以下步骤,(1)取酿造酱油样品a,并进行脱色,脱色后为脱色后的酱油样品b;(2)取含有v2ml酱油样品a的脱色后的酱油样品b,并向其滴加指示剂,此时溶液显示为指示剂的酸色,然后用胶头滴管滴加0.050mol/l的naoh至溶液颜色显示为指示剂的酸色和碱色的混合色,且半分钟不褪色;然后加入中性甲醛,把所取酱油样品b中的氨基酸态氮完全转化为酸性物质(-cooh),此时溶液显示为指示剂的酸色;然后加入v3ml的一元强碱溶液,轻轻摇晃后观察溶液颜色,此时,溶液显示为指示剂的酸色或溶液酸碱“等量点”时显示指示剂的酸色和碱色的混合色,且半分钟不褪色则该样品为符合一定级别的合格酿造酱油;若溶液显示为指示剂的碱色且半分钟不褪色则样品为不符合一定级别的不合格酿造酱油;一元强碱溶液的浓度与酱油样品a所在等级的最低浓度相等,且v2=v3。
一元强碱溶液为氢氧化钠溶液、氢氧化钾溶液或氢氧化锂溶液。
酱油样品a为酿造酱油原样,酱油原样为市场售卖或工厂生产的酱油产品;酱油原样为李锦记味极鲜、厨邦海鲜酱油/生抽、海天酱油老抽王、李锦记精选生抽、李锦记锦珍老抽、加加草菇老抽、加加金标生抽、鲁花自然鲜酱香酱油。
指示剂为紫薯色素。
紫薯色素的制备方法为:取紫薯,并研磨成糊状,按料液比1:5加入蒸馏水,移入100ml比色管中,用超声波辅助提取,超声波功率为500w,提取温度为55℃,时间为15-20min,然后过滤到得到紫薯色素;所用蒸馏水的ph为6.8。
所述步骤(1)中酿造酱油进行脱色为:用移液枪移取1.00ml酱油原样样品于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,得到的过滤后的澄清液为脱色后的酱油样品b。
所述步骤(2)中,用移液枪取1.00ml酱油样品b于10ml比色管中,滴加2至3滴天然紫薯色素,此时溶液为红色或红橙色,用胶头滴管逐滴滴加0.050mol/lnaoh标准溶液至颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色或红橙色。
紫薯色素的酸色显示为红色或红橙色,紫薯色素的碱色显示为灰蓝色、绿土色或黄棕色,紫薯色素的红橙色与灰蓝色的混合色为橙灰色;按所述步骤方法鉴别时,被鉴别样品溶液中余剩的酸性物质的量,红色大于红橙色,红橙色大于橙灰色;溶液中余剩的一元强碱量,黄棕色大于绿土色,绿土色大于灰蓝色。
下面结合具体实施例对本发明进行具体描述,有必要在此指出的是本实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,该领域的技术熟练人员可以根据上述本发明的内容做出一些非本质的改进和调整。本申请中,轻轻摇晃后观察溶液颜色,轻轻摇晃为滴定过程中常规的摇晃程度。
一、紫薯色素的制备(用蒸馏水或纯净水)
将10g-15g新鲜紫薯(紫薯样品2019年10月30日购自郑州市二七区马寨镇联合一百超市,购置时非常新鲜,色泽呈紫色至深紫色)切成小片状,置于研钵中研磨呈糊状后,按料液比1:5加入蒸馏水,移入100ml比色管中,用超声波(功率为500w)辅助提取,提取温度为55℃,时间为10-20min。然后过滤到滴瓶中,即为紫薯色素。所用蒸馏水的ph为6.8。
取12个洁净100ml小烧杯,各加约40ml蒸馏水,在酸度计上小心滴加0.10mol/l盐酸或0.10mol/l氢氧化钠溶液,调节至ph分别为2.00、3.00、4.00、5.00、6.00、7.00、8.00、9.00、10.00、11.00、12.00和13.00,然后各滴加4滴紫薯色素过滤液,摇匀后,分别移入25.00毫升比色管中,观察紫薯色素在各不同ph值溶液中的颜色变化,如图1,自左至右:颜色由红色(ph2、3)、红橙色(ph4、5、6、7)、橙灰色(ph8)、灰蓝色(ph9)、绿土色(ph10、11)至黄棕色(ph12、13)。
二、naoh标准溶液的配制、标定、及浓度调节
1、浓度的设定
氨基酸为中性内盐,加入中性甲醛时,呈现碱性的氨基(-nh2)与甲醛结合,碱性消失,而呈现酸性羧基(-cooh),可与强碱标准溶液等物质量的反应,尤其适用于发酵液中氨基态氮的测定。每个氨基(-nh2)中含有一个氮。无论是高盐稀态发酵酱油,还是低盐固态发酵酱油,各级产品均以氨基酸态氮(以氮计)含量为主要理化指标,在处理样品后加入中性甲醛转化氨基酸态氮时,产品要求氮的最低浓度即是要求的氨基酸的最低浓度,也是转化后羧基的最低浓度,因此,各级合格产品中的羧基(-cooh)的最低浓度应为:
三级:
二级:
一级:
特级:
所以,根据“等物质量的反应”原则,本发明配制相应级别同浓度的naoh(或koh)强碱标准溶液,与样品中的羧基(-cooh)发生“等物质量的反应”,反应完成后,如果样品羧基剩余,则是相应级别的合格产品;若羧基不足,强碱剩余,则酿造酱油是相应级别的不合格产品。若所取样品中羧基(-cooh)的物质的量与加入的强碱的物质的量正好相等,中和反应后均不剩余,正好达到所要求级别最低指标的理论终点,溶液呈现的是指示剂红橙色和灰蓝色的混合色,即橙灰色,这种情况极为罕见。用脱色剂脱除酱油的干扰颜色后,用合适的指示剂可以快速甄别和判断。
2、不同浓度naoh标准溶液的配制、标定和调节
先用电子天平称取需要的naoh(a.r)用量并溶解、定容。用烘至恒质量的邻苯二甲酸氢钾基准物质进行标定,进行4次平行标定,检验无可疑值后取平均值,若浓度低于目标浓度,计算并添加所需固体naoh(a.r);若浓度高于目标浓度,计算并添加所需蒸馏水。
(1)0.2857mol/lnaoh标准溶液的配制、标定和调节
用电子天平快速称取naoh(a.r,分析纯)5.714g于小烧杯中,用80ml左右蒸馏水溶解,冷至室温后,定量移入500ml容量瓶中,用蒸馏水定容至刻度,混匀、备用。
在分析天平上准确称取105℃烘至恒质量的邻苯二甲酸氢钾基准试剂0.6g-1.2g(准确到0.0001g)于洁净的锥形瓶中,加蒸馏水约40ml使之完全溶解,加入本申请制备的紫薯色素4滴,用处理好的碱式滴定管中的上述氢氧化钠溶液进行标定,滴定至红橙色刚好消失变为橙灰色半分钟不褪色为终点,记录消耗氢氧化钠溶液的体积v1(毫升),同时取40ml蒸馏水进行空白试验和颜色比对,按下式计算浓度:
c(naoh)=(m×1000÷204.22)/(v1-v0)
其中,c(naoh):氢氧化钠标准溶液的物质的量浓度,mol/l;m为准确称取的邻苯二甲酸氢钾基准试剂的质量,g;204.22为邻苯二甲酸氢钾的摩尔质量,g/mol;v0为空白液所消耗的氢氧化钠溶液的体积(ml);v1为标定质量为m的邻苯二甲酸氢钾基准试剂所消耗的氢氧化钠溶液的体积(ml)。
做4次平行标定,检验无可疑值后取平均值,若浓度低于0.2857mol/l,计算并添加所需的固体naoh;若浓度高于0.2857mol/l,计算并添加所需蒸馏水,直至重新标定后达到目标浓度。
也可以配制0.2857mol/l的氢氧化钾标准溶液进行工作,本发明选择价格较便宜的氢氧化钠标准溶液,投入成本相对降低。
(2)0.3929mol/lnaoh标准溶液的配制、标定和调节
用电子天平快速称取naoh(分析纯)7.858g于小烧杯中,用80ml左右蒸馏水溶解,冷至室温后,定量移入500ml容量瓶中,用蒸馏水定容至刻度,混匀、备用。
在分析天平上准确称取105℃烘至恒质量的基准试剂邻苯二甲酸氢钾0.8g-1.6g(准确到0.0001g)于洁净的锥形瓶中,加蒸馏水约40ml使之完全溶解,加入本申请制备的紫薯色素4滴,用处理好的碱式滴定管中的上述氢氧化钠溶液进行标定,滴定至由红橙色刚好消失变为橙灰色,半分钟不褪色为终点,记录消耗氢氧化钠溶液的体积v1(ml),同时取40ml蒸馏水进行空白试验和颜色比对,按下式计算浓度:
c(naoh)=(m×1000÷204.22)/(v1-v0)
其中,c(naoh):氢氧化钠标准溶液的物质的量浓度,mol/l;m为准确称取的邻苯二甲酸氢钾基准试剂的质量,g;204.22为邻苯二甲酸氢钾的摩尔质量,g/mol;v0为空白液所消耗的氢氧化钠溶液的体积(ml);v1为标定质量为m的邻苯二甲酸氢钾基准试剂所消耗的氢氧化钠溶液的体积(ml)。
做4次平行标定,检验无可疑值后,取平均值,若浓度低于0.3929mol/l,计算并添加所需的固体naoh;若浓度高于0.3929mol/l,计算并添加所需蒸馏水,直至重新标定后达到目标浓度。
也可以配制0.3929mol/l的氢氧化钾标准溶液进行工作,本发明选择价格较便宜的氢氧化钠标准溶液,投入成本相对降低。
(3)0.4286mol/lnaoh标准溶液的配制、标定和调节
用电子天平快速称取naoh(分析纯)8.5720克于小烧杯中,用80ml左右蒸馏水溶解,冷至室温后,定量移入500ml容量瓶中,用蒸馏水定容至刻度,混匀、备用。
在分析天平上准确称取105℃烘至恒质量的基准试剂邻苯二甲酸氢钾0.8g-1.8g(准确到0.0001g)于洁净的锥形瓶中,加蒸馏水约40ml使之完全溶解,加入本申请制备的紫薯色素4滴,用处理好的碱式滴定管中的上述氢氧化钠溶液进行标定,滴定至红橙色刚好消失变为橙灰色,半分钟不褪色为终点,记录消耗氢氧化钠溶液的体积v1(ml),同时取40ml蒸馏水进行空白试验和颜色比对,按下式计算浓度:
c(naoh)=(m×1000÷204.22)/(v1-v0)
其中,c(naoh)为氢氧化钠标准溶液的物质的量浓度,mol/l;m为准确称取的邻苯二甲酸氢钾基准试剂的质量,g;204.22为邻苯二甲酸氢钾的摩尔质量,g/mol;v0为空白液所消耗的氢氧化钠溶液的体积(ml);v1为标定质量为m的邻苯二甲酸氢钾基准试剂所消耗的氢氧化钠溶液的体积(ml)。
做4次平行标定,检验无可疑值后,取平均值,若浓度低于0.4286mol/l,计算并添加所需的固体naoh;若浓度高于0.4286mol/l,计算并添加所需蒸馏水,直至重新标定后达到目标浓度。
也可以配制0.4286mol/l的氢氧化钾标准溶液进行工作,此发明选择价格较便宜的氢氧化钠标准溶液,投入成本相对降低。
(4)0.5000mol/lnaoh标准溶液的配制、标定和调节
用电子天平快速称取naoh(分析纯)10.00g于小烧杯中,用80ml左右蒸馏水溶解,冷至室温后,定量移入500ml容量瓶中,用蒸馏水定容至刻度,混匀、备用。
在分析天平上准确称取105℃烘至恒质量的基准试剂邻苯二甲酸氢钾1.0g-2.0g(准确到0.0001g)于洁净的锥形瓶中,加蒸馏水约40ml使之完全溶解,加入本申请制备的紫薯色素4滴,用处理好的碱式滴定管中的上述氢氧化钠溶液进行标定,滴定至红橙色刚好消失变为橙灰色,半分钟不褪色为终点,记录消耗氢氧化钠溶液的体积v1(ml),同时取40ml蒸馏水进行空白试验和颜色比对,按下式计算浓度:
c(naoh)=(m×1000÷204.22)/(v1-v0)
其中,c(naoh)为氢氧化钠标准溶液的物质的量浓度,mol/l;m为准确称取的邻苯二甲酸氢钾基准试剂的质量,g;204.22为邻苯二甲酸氢钾的摩尔质量,g/mol;v0为空白液所消耗的氢氧化钠溶液的体积(ml);v1为标定质量为m的邻苯二甲酸氢钾基准试剂所消耗的氢氧化钠溶液的体积(ml)。
做4次平行标定,检验无可疑值后,取平均值,若浓度低于0.5000mol/l,计算并添加所需的固体naoh;若浓度高于0.5000mol/l,计算并添加所需蒸馏水,直至重新标定后达到目标浓度。
也可以配制0.5000mol/l的氢氧化钾标准溶液进行工作,此发明选择价格较便宜的氢氧化钠标准溶液,投入成本相对降低。
(5)0.5714mol/l的naoh配制、标定和调节
用电子天平快速称取naoh(分析纯)11.428g于小烧杯中,用80ml左右蒸馏水溶解,冷至室温后,定量移入500ml容量瓶中,用蒸馏水定容至刻度,混匀、备用。
在分析天平上准确称取105℃烘至恒质量的基准试剂邻苯二甲酸氢钾1.2g-2.5g(准确到0.0001g)于洁净的锥形瓶中,加蒸馏水约40ml使之完全溶解,加入本申请制备的紫薯色素4滴,用处理好的碱式滴定管中的上述氢氧化钠溶液进行标定,滴定至红橙色刚好消失变为橙灰色半分钟不褪色为终点,记录消耗氢氧化钠溶液的体积v1(ml),同时取40ml蒸馏水进行空白试验和颜色比对,按下式计算浓度:
c(naoh)=(m×1000÷204.22)/(v1-v0)
其中,c(naoh)为氢氧化钠标准溶液的物质的量浓度,mol/l;m为准确称取的邻苯二甲酸氢钾基准试剂的质量,g;204.22为邻苯二甲酸氢钾的摩尔质量,g/mol;v0为空白液所消耗的氢氧化钠溶液的体积(ml);v1为标定质量为m的邻苯二甲酸氢钾基准试剂所消耗的氢氧化钠溶液的体积(ml)。
做4次平行标定,检验无可疑值后取平均值,若浓度低于0.5714mol/l,计算并添加所需的固体naoh;若浓度高于0.5714mol/l,计算并添加所需蒸馏水,直至重新标定后达到目标浓度。
也可以配制0.5714mol/l的氢氧化钾标准溶液进行工作,此发明选择价格较便宜的氢氧化钠标准溶液,投入成本相对降低。
三、中性甲醛的配制
36%-38%的分析纯甲醛溶液中滴加4-6滴本申请制备的紫薯色素,向甲醛溶液中逐滴滴加naoh溶液,边滴边轻轻摇动,滴至甲醛溶液由红橙色消失,刚好变为橙灰色,密闭保存,备用。
四、酱油的脱色处理及效果
实验仪器及试剂:uv-2800ah型紫外可见分光光度计(尤尼柯仪器有限公司)、200目食品糖类活性炭(罗恩试剂)。
实验步骤:
(1)取1.00ml李锦记味极鲜原酱油于洁净试管中,加入9.00ml去离子水,混匀、备用,此为待扫描试液。
(2)空白液的制备:取1.00ml李锦记味极鲜原酱油于洁净试管中,加入9.00ml去离子水,加入0.40g活性炭处理,静置2min,用漏斗过滤,以滤液作为空白(以排除色素以外的其它因素对吸光度产生影响)。
(3)吸收光谱的绘制:以稀释10倍的样品液为试液,以空白样品液为对照,用1cm的比色皿扫描200-800nm间试液的吸收光谱表明:李锦记味极鲜原酱油的最大波长λmax=336.0nm。
(4)脱色率的研究:取10倍稀释酱油各20ml分置于7个50.00ml比色管中,分别依次加入0.12g、0.16g、0.20g、0.24g、0.28g、0.30g、0.32g活性炭轻轻震荡,静置1min-2min,用滤纸过滤。以空白样品液为对照,以336.0nm为入射光,用1cm的比色皿测定各试液的吸光度。按下式计算出样品的脱色率:
脱色率,%=(a0-a)÷a0×100%(3)
式中,a0为10倍稀释酱油样品未脱色时测定的吸光度,a为10倍稀释酱油样品加入不同量活性炭脱色后,测定过滤液的吸光度。结果如图2。由图2可知,加入0.30g活性炭时脱色率为92.56%,可达到较佳脱色效果。
称取0.30g活性炭分别静置2min、4min、6min后过滤,按上述方法测定吸光度并计算脱色率。结果如图3。由图3可知,脱色2min至6min效果已令人满意,为提高工作效率,本发明脱色时间选择为2min。
五、国标法电位滴定法测定
(1)用氢氧化钾标准溶液电位滴定法测定
操作步骤:吸5.00ml酱油于100ml容量瓶中,加水至刻度线,混匀后吸取20ml,置于100ml烧杯中,加入60ml水。用氢氧化钾标准溶液c(koh=0.050mol/l)滴定至离子计ph=8.2,记下所消耗的koh体积。加入20ml中性甲醛,混匀,记录此时的ph。再用koh继续滴定至ph=8.2,记录所消耗的koh体积。
同时取20ml水,先用0.050mol/lkoh调至ph=8.2,再加入20ml中性甲醛,用0.050mol/lkoh滴定至ph=8.2,记录所消耗的koh体积,作为试剂的空白实验。
试样中氨基酸态氮含量的计算公式为:
式中:
x—试样中氨基酸态氮含量,g/100ml;
v1—测定用试样稀释液加入甲醛后消耗koh的体积,ml;
v0—试剂空白实验加入甲醛后消耗koh的体积,ml;
v—试样稀释液取用量,ml;
c—koh的物质的量浓度,mol/l;
0.014——与1.00mmol(毫摩尔)koh相当的氮的质量,g。
计算结果保留三位有效数字。
(2)用氢氧化钠标准溶液电位滴定法测定
操作步骤:吸5.00ml酱油于100ml容量瓶中,加水至刻度线,混匀后吸取20ml,置于100ml烧杯中,加入60ml水。用氢氧化钠标准溶液c(naoh=0.050mol/l)滴定至离子计ph=8.2,记下所消耗的naoh体积。加入20ml没有调节至中性的甲醛,混匀,记录此时的ph。再用naoh继续滴定至ph=9.2,记录所消耗的naoh体积。
同时取20ml水,先用0.050mol/lnaoh调至ph=8.2,再加入20ml没有调节至中性的甲醛,用0.050mol/lnaoh滴定至ph=9.2,记录所消耗的naoh体积,作为试剂的空白实验。
试样中氨基酸态氮含量的计算公式为:
式中:
x—试样中氨基酸态氮含量,g/100ml;
v1—测定用试样稀释液加入甲醛后消耗强碱标准溶液的体积,ml;
v0—试剂空白实验加入甲醛后消耗强碱标准溶液的体积,ml;
v—试样稀释液取用量,ml;
c—强碱标准溶液的物质的量浓度,mol/l;
0.014——与1.00mmol(毫摩尔)强碱相当的氮的质量,g。
计算结果保留三位有效数字。
(3)、样品对照测定结果及分析如下表表1。
国标法检测值总是略大于本项目的检测值,主要原因是国标法加入甲醛转化氨基酸态氮前,试液的ph处理为8.2,加入甲醛后用naoh标准溶液滴定至ph9.2为终点,违背了分析化学定量分析中的“等物质的量转化和反应原则”,使测定结果偏高。
六、原酿造酱油按国标法第一法酸度计法的测定结果
按国标法gb5009.235-2016《食品中氨基酸态氮含量的测定》的第一法酸度计法对样品进行测定,和试剂管溶液颜色、商品标注值进行比较,结果如表2。
由表2可知,国标法样品测定结果均高于商品标注值。
七、酿造酱油氨基酸态氮含量是否符合一定级别的快速检测方法及结果验证
(一)酱油原样的氨基酸态氮含量是否符合一定级别的快速检测方法及结果验证
(1)特级酱油(氨基酸态氮含量≥0.80g/100ml)
实验试剂:0.5714mol/lnaoh溶液、中性甲醛
操作步骤:用移液枪移取1.00ml酱油于25ml比色管中,加9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,用移液枪移取1.00ml过滤澄清液于10ml比色管中,滴加2至3滴紫薯天然色素,此时溶液为红色,用胶头滴管滴加0.050mol/lnaoh至溶液颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色,用移液枪加0.5714mol/lnaoh溶液100μl,轻轻摇晃后观察溶液颜色,此时溶液为红色、红橙色或橙灰色且半分钟不褪色,则产品为特级酿造酱油,若为灰蓝色、绿土色或黄棕色且半分钟不褪色,则产品质量不符合特级酿造酱油的要求。
用中性甲醛将氨基酸态氮等物质量的转化出酸(-cooh),所以,溶液的酸度大大增加,色素呈现为红色。然后加入相应级别产品所需要的强碱后观察溶液颜色,进行快速鉴别。以下相同。2.00ml中性甲醛可把所取100μl原酱油样品中的氨基酸态氮完全转化为等物质量的酸性物质(-cooh)。
(2)一级酱油(氨基酸态氮含量≥0.70g/100ml)
实验试剂:0.5000mol/lnaoh溶液、中性甲醛
操作步骤:用移液枪移取1.00ml酱油于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,用移液枪移取1.00ml过滤澄清液于10ml比色管中,滴加2至3滴天然紫薯色素,此时溶液为红色,用胶头滴管滴加0.050mol/lnaoh至溶液颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色,用移液枪加入0.5000mol/lnaoh溶液100μl,轻轻摇晃后观察溶液颜色,若溶液仍为红色、红橙色或橙灰色且半分钟不褪色,则产品为一级发酵酿造酱油,若为灰蓝色、绿土色或黄棕色且半分钟不褪色,则产品不符合一级发酵酿造酱油的要求。
(3)二级酱油(高盐稀态发酵酱油氨基酸态氮含量≥0.55g/100ml)
实验试剂:0.3929mol/lnaoh标准溶液、中性甲醛
操作步骤:用移液枪移取1.00ml酱油于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,取1.00ml澄清液于10ml比色管中,滴加2至3滴天然紫薯色素,此时溶液为红色,用胶头滴管滴加0.050mol/lnaoh溶液至颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色,用移液枪加0.3929mol/lnaoh溶液100μl,轻轻摇晃后观察溶液颜色,此时溶液仍为红色、红橙色或橙灰色且半分钟不褪色,则产品为二级高盐稀态发酵酱油,若为灰蓝色、绿土色或黄棕色且半分钟不褪色,则产品不符合二级高盐稀态发酵酱油的要求。
(4)二级酱油(低盐固态发酵酱油氨基酸态氮含量≥0.60g/100ml)
实验试剂:0.4286mol/lnaoh溶液、中性甲醛
操作步骤:用移液枪移取1.00ml酱油于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,取1.00ml澄清液于10ml比色管中,滴加2至3滴天然紫薯色素,此时溶液为红色,用胶头滴管滴加0.050mol/lnaoh溶液至颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色,用移液枪加0.4286mol/lnaoh标准溶液100μl,轻轻摇晃后观察溶液颜色,此时溶液仍为红色、红橙色或橙灰色且半分钟不褪色,则产品为二级低盐固态发酵酱油,若为灰蓝色、绿土色或黄棕色且半分钟不褪色,则产品不符合二级低盐固态发酵酱油的要求。
(5)三级酱油(氨基酸态氮含量≥0.40g/100ml)
实验试剂:0.2857mol/lnaoh标准溶液、中性甲醛
操作步骤:用移液枪移取1.00ml酱油于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,用移液枪取1.00ml澄清液于10ml比色管中,滴加2至3滴天然紫薯色素,此时溶液为红色,用胶头滴管滴加0.050mol/lnaoh标准溶液至颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色,用移液枪加入0.2857mol/lnaoh标准溶液100μl,轻轻摇晃后观察溶液颜色,此时溶液仍为红色、红橙色或橙灰色且半分钟不褪色,则产品为三级发酵酱油,若为灰蓝色、绿土色或黄棕色且半分钟不褪色,则产品不符合三级发酵酱油的要求,即不符合国家相关标准。
结果验证:本申请提供的快速检测方法得到的结果与国标法得到的结果完全吻合,如表3。
(二)稀释后的酿造酱油原样的氨基酸态氮含量是否合格的快速检测的方法及结果验证
将酱油原样按照一定的比例用蒸馏水稀释后作为样品,根据国家食品安全标准gb5009.235-2016《食品中氨基酸态氮含量的测定》的第一法为酸度计法,并按照(1)计算结果,平行测定3次,检验无可疑值后取平均值报告,不同比例稀释后样品按国标法测定的结果如表4、表5。
1、酿造酱油原样稀释一倍样品的制备和测定
用移液管移取12.50ml酱油原样于25.00ml容量瓶中,用水(ph=6.80)的怡宝纯净水稀释至刻度,混匀备用。按国标法gb5009.235-2016《食品中氨基酸态氮含量的测定》的第一法酸度计法对样品进行3次平行测定,检验无可疑值后,取平均值报告。
同时,用移液枪移取酱油原样稀释一倍的样品1.00ml于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻璃棒轻轻搅动脱色,2min后过滤,用移液枪取1.00ml脱色后的澄清液于10ml比色管中,滴加2至3滴本申请制备的紫薯色素,此时溶液为红色,用胶头滴管逐滴滴加0.050mol/lnaoh标准溶液至颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色,用移液枪加各级别要求对应浓度的上述naoh标准溶液100μl,轻轻摇晃后观察溶液颜色,此时溶液仍为红色、红橙色或橙灰色且半分钟不褪色,则产品为合格级别的酿造酱油,若为灰蓝色、绿土色或黄棕色且半分钟不褪色,则产品不符合对应级别酿造酱油的要求,即不符合国家相关标准,试剂管溶液颜色和国标法测定结果如表4。
2、酱油原样稀释五倍后样品的制备和测定
用移液管移取5.00ml酱油原样于25.00ml容量瓶中,用水(ph=6.80)的怡宝纯净水稀释至刻度,混匀备用。按国标法gb5009.235-2016《食品中氨基酸态氮含量的测定》的第一法酸度计法对样品进行3次平行测定,检验无可疑值后,取平均值报告。
同时,用移液枪移取酱油原样稀释五倍后的样品1.00ml于25ml比色管中,加入9.00ml水稀释,加入0.30g活性炭,用玻棒轻轻搅动脱色,2min后过滤,用移液枪取1.00ml脱色后的澄清液于10ml比色管中,滴加2至3滴本申请制备的紫薯色素,此时溶液为红色,用胶头滴管滴加0.050mol/lnaoh标准溶液至颜色为橙灰色,加入2.00ml中性甲醛,此时溶液为红色,用移液枪加各级别要求对应浓度的上述naoh标准溶液100μl,轻轻摇晃后观察溶液颜色,此时溶液仍为红色、红橙色或橙灰色且半分钟不褪色,则产品为合格级别的酿造酱油,若为灰蓝色、绿土色或黄棕色且半分钟不褪色,则产品不符合对应级别酿造酱油的要求,即不符合国家相关标准,试剂管溶液颜色和国标法测定结果如表5。
本发明均根据各级别酿造酱油的氨基酸态氮的合格指标,按照“等物质量的反应”原则,设定加入标准氢氧化钠溶液的体积为100微升,即0.10ml原酿造酱油进行快速鉴别。按所述步骤方法鉴别时,样品溶液中余剩的酸性物质(-cooh)的量,红色大于红橙色,红橙色大于橙灰色;溶液中余剩的一元强碱(-oh)量,黄棕色大于绿土色,绿土色大于灰蓝色。
本发明根据酿造酱油不同级别理化指标氨基酸态氮合格的最低要求,科学设计一元强碱的浓度。依据等物质的量反应原则,当所取酱油原样品的体积确定以后,加入同体积的一元强碱溶液,如果所取酱油样品中氨基酸态氮的浓度大于一元强碱溶液的浓度,用中性甲醛转化氨基酸态氮生成的酸性物质(-cooh)的浓度也大于一元强碱溶液的浓度,反应完成后,酸性物质余剩,该产品为合格产品,余剩的酸越多,产品质量相对越好;如果所取酿造酱油等物质量转化出酸性物质的浓度正好等于一元强碱溶液的浓度,反应完成后,恰好到达符合相应级别酱油最低要求的理论终点,且酸、碱物质均不余剩,酿造酱油是符合最低指标要求的合格产品;如果所取酱油转化出的酸性物质(-cooh)的浓度小于一元强碱溶液的浓度,反应完成后,强碱余剩,-cooh不足,该酿造酱油中的氨基酸态氮低于相应级别的最低要求,为不符合相应级别的产品。此结论可用合适的指示剂加以快速鉴别。
一元强碱滴定弱酸,滴定突跃总是落在碱性区域。突跃范围的大小与酸碱的浓度和弱酸的ka的大小有关,如:用0.01000mol/l的氢氧化钠滴定0.01000mol/l的醋酸,滴定突跃在ph7.7至ph8.7,理论终点为ph8.23(可参阅有关《分析化学》书籍);紫薯色素的颜色为红色(ph2、3)、红橙色(ph4、5、6、7)、橙灰色(ph8)、灰蓝色(ph9)、绿土色(ph10、11)至黄棕色(ph12、13)。
因此,酿造酱油溶液,按本发明技术操作,紫薯色素颜色结果为红色、红橙色,反应结束后,各级产品位于符合相应级别酿造酱油最低氨基酸态氮要求的理论终点前,酸性物质(-cooh)剩余,可立刻判别为合格样品;如果紫薯色素颜色为橙灰色,反应正好到达符合相应级别酱油最低氨基酸态氮要求的“理论终点”,溶液中的酸、碱物质的量正好相等,均不过量,可判别酿造酱油刚好为相应级别的合格样品;如果呈现为灰蓝色、绿土色或黄棕色,反应已经超过了最低氨基酸态氮要求的理论终点,强碱过量,此为不合格样品。
以上所说,如果紫薯色素颜色为橙灰色,反应正好到达符合各级酱油最低氨基酸态氮所要求的理论终点,溶液中酸、碱物质的量正好相等,均不过量,可判别样品刚好为合格;反应结束在理论终点附近,这种情况极少见到,由此造成的相对误差不会大于0.1%,即测定的准确度大于99.9%(滴定突跃的范围是理论终点前后相对误差为-0.1%到+0.1%的区间),这是滴定反应和测定所允许的范围。在实际操作鉴别的大量具体样品中,尚没有见到过实际样品中紫薯色素的橙灰色或灰蓝色色调。因生产厂家的产品中,氨基酸态氮正好按照国标要求的最低限设计工艺的几乎为零,截至目前还没有见到。
另外补充说明:由于溶液的酸度即ph值决定酸碱指示剂的存在形态、以及酸色和碱色形态的比值,也就决定了指示剂的具体颜色,即溶液的酸度决定酸碱指示剂的颜色。而同一颜色的深浅与颜色物质的浓度有关,即与单位体积内相关颜色物质的量有关,即指示剂的浓度决定颜色的深浅。同样酸度下,溶液的颜色深浅与加入指示剂的量呈正比。如果同样体积的样品,鉴别中只加入一滴紫薯色素,颜色会偏浅一些,但不影响结论的判断。因为加入指示剂的量,通常不会影响溶液的酸度,不会影响指示剂的存在形态和比值。加入的紫薯色素少,单位体积内紫薯色素的有色质点少,同一颜色就会变淡些,加入的紫薯色素多,同一颜色就会深一些,但它们仍然是各自的红色(含深红、红色、浅红色、橙红色、粉红色、微红色等)、各自的绿色(含绿色、深绿色、草绿色、绿土色、浅绿色、淡绿色等)和各自的黄色(含棕黄色、深黄色、黄色、淡黄色、绿黄色等),只是各自颜色的深浅程度不同而已,只要指示剂和溶液的酸度固定不变,也不受溶液、空气中其它物质和因素的干扰,颜色通常不会发生根本变化,绿色不会变为黄色,也不会变为红色,反之一样。只要能正确判断,获得符合实际的结果即可。
以上所述的仅是本发明的优选实施方式,但本发明的保护范围并不局限于此,应当指出,对于本领域的及任何熟悉本技术领域的技术人员来说,在不脱离本发明整体构思前提下,根据本发明的技术方案及其发明构思加以等同替换或改变,及作出的若干改变和改进,这些也应该视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!