一种发泡剂产品纯度及其杂质含量的检测方法与流程
本发明涉及有机发泡剂纯度分析
技术领域:
,具体涉及一种发泡剂产品纯度及其杂质含量的检测方法。
背景技术:
:发泡剂h为有机化学发泡剂,主要成分为n,n-二亚硝基五亚甲基四胺,加热(>205℃)分解产生氮气,具有发气量大、鼓泡效率高等特点,可用作聚氯乙烯及其共聚物、聚烯烃、聚苯乙烯、聚酰胺、聚酯、酚醛树脂、聚偏二氯乙烯、聚硅氧烷、聚氯丁二烯、乙烯和丙烯共聚物、聚氧化乙烯及其弹性体的发泡剂,是广泛用于生产具有质轻、隔热、良好机械阻尼特性的橡胶和塑料系列产品的助剂。核磁共振氢谱技术主要用于有机化合物的结构测定,同时也是有机化合物定量分析的重要手段,其基本原理是1h-nmr中的共振峰面积与该共振峰包含氢原子个数成正比,该法的优点是无需引入任何校正因子、速度快、简单准确,目前很多国家和地区药典已收录定量核磁分析方法。目前尚未见到通过核磁共振氢谱来确定检测发泡剂h中的n,n-二亚硝基五亚甲基四胺的含量的技术。技术实现要素:目前发泡剂h是以六次亚甲基四胺和亚硝酸钠为原料,在低温和酸性条件下反应合成的,在生产过程中主要副产物为无机盐和三亚硝基三亚甲基三胺(ttt),无机盐可以通过水洗去除,而三亚硝基三亚甲基三胺与n,n-二亚硝基五亚甲基四胺由于相同的官能团具有类似的物理、化学性质,难以将两者彻底分离,只能通过优化工艺条件和参数,抑制ttt的产生。然而发泡剂h产品纯度主要依靠检测样品中氮元素含量的经验方法,该方法不仅不科学,而且难以确定副产物三亚硝基三亚甲基三胺的含量。本发明针对上述情况,为了解决传统检测发泡剂h方法中不能精确区分n,n-二亚硝基五亚甲基四胺和三亚硝基三亚甲基三胺的不足之处,本发明提供一种发泡剂h产品纯度的检测方法,该方法利用核磁共振氢谱内标法,具有内标物廉价易得、简单准确及重复性好等优点。现有技术中未有采用核磁共振氢谱来区分检测n,n-二亚硝基五亚甲基四胺、三亚硝基三亚甲基三胺等组分的含量的检测方法。具体来说,本发明提出了如下技术方案:本发明提供了一种发泡剂产品纯度及杂质含量的检测方法,其特征在于,包含下述步骤:(1)称取样品,干燥,计算样品中的水分w%;(3)焙烧样品,计算样品中的灰分s%;(3)称取发泡剂和内标物置于样品瓶中,加入氘代溶剂溶解后,进行核磁共振氢谱测试;(4)确定步骤(3)核磁共振氢谱中化合物n,n-二亚硝基五亚甲基四胺和内标物的特征峰,设定核磁共振氢谱测定条件,测定其相对积分面积分别为ah和as,将数据代入下式得到测定n,n-二亚硝基五亚甲基四胺质量分数:其中,ph为n,n-二亚硝基五亚甲基四胺的质量分数;ps为内标物的纯度;ah为n,n-二亚硝基五亚甲基四胺的峰面积;as为内标物的峰面积;nh为1摩尔n,n-二亚硝基五亚甲基四胺特征信号的官能团上共振核的数目;ns为1摩尔内标物特征信号的官能团上共振核的数目;mh为n,n-二亚硝基五亚甲基四胺的摩尔质量;ms为内标物的摩尔质量;mh为n,n-二亚硝基五亚甲基四胺的质量;ms为内标物的质量;(5)采用步骤(4)相同的操作测定并计算发泡剂中化合物三亚硝基三亚甲基三胺的质量分数。优选的,其中,步骤(3)中所述内标物选自对二氯苯或对二硝基苯,优选的,所述内标物为高纯品或标准品。优选的,其中,步骤(1)或步骤中(2)所用样品的质量为3~4g;优选地,步骤(3)中所用样品质量为30~60mg。优选的,其中,所述发泡剂产品中水分为0.20-0.29wt%,灰分为0.060-0.083wt%;优选地,三亚硝基三亚甲基三胺的含量为0.8-2.0wt%。优选的,其中,步骤(3)中氘代溶剂的用量为2.0-8.0ml;优选地,步骤(4)中内标物的质量为10-25mg。优选的,其中,步骤(4)中核磁共振氢谱测试条件包括:核磁波谱仪共振频率为400~600mhz;优选地,温度为20~35℃;优选地,脉冲角度为90°;优选地,延迟时间20~40s;优选地,采样次数为32~128次。优选的,其中,步骤(1)中进行干燥的温度为70~100℃,优选干燥时间为2~4小时。优选的,其中,步骤(2)中进行焙烧的温度为500~800℃,优选焙烧时间为2~4小时。优选的,其中,所述氘代溶剂为重氢二甲基亚砜(dmso-d6),氘代氯仿和氘代乙腈中的一种或两种以上;优选为重氢二甲基亚砜(dmso-d6)。本发明还提供了所述的方法在有机发泡剂纯度分析
技术领域:
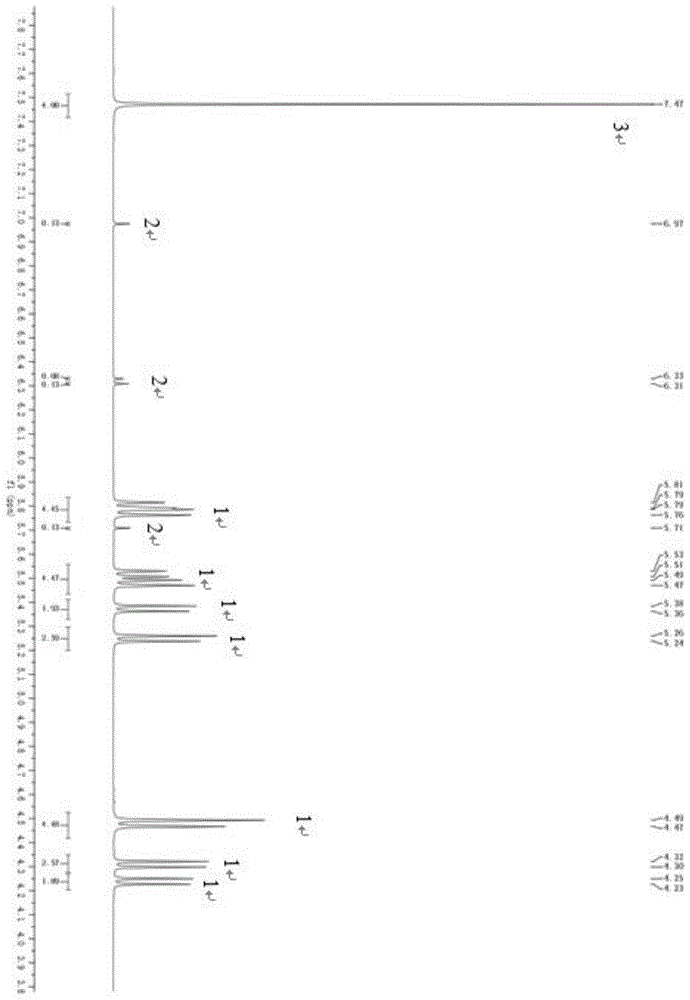
的应用。本发明的有益效果包括:本发明利用1h-nmr技术,选择廉价稳定的内标物,可以快速、准确分析发泡剂h样品中n,n-二亚硝基五亚甲基四胺和三亚硝基三亚甲基三胺的含量,可以替代传统经验分析方法。附图说明图1为实施例1-1的核磁共振氢谱图,其中,图中编号1为n,n-二亚硝基五亚甲基四胺的化学位移特征峰,编号2为三亚硝基三亚甲基三胺的化学位移特征峰,编号3为内标物的化学位移特征峰。具体实施方式本发明针对发泡剂产品纯度传统检测方法的缺陷,提供了一种能够快速、准确检测出n,n-二亚硝基五亚甲基四胺和三亚硝基三亚甲基三胺含量的核磁共振氢谱测量方法。本发明提供了一种发泡剂产品纯度及杂质含量的检测方法,其特征在于,包含下述步骤:(1)称取样品,干燥,计算样品中的水分w%;(4)焙烧样品,计算样品中的灰分s%;(3)称取发泡剂和内标物置于样品瓶中,加入氘代溶剂溶解后,进行核磁共振氢谱测试;(4)确定步骤(3)核磁共振氢谱中化合物n,n-二亚硝基五亚甲基四胺和内标物的特征峰,设定核磁共振氢谱测定条件,测定其相对积分面积分别为ah和as,将数据代入下式得到测定n,n-二亚硝基五亚甲基四胺质量分数:其中,ph为n,n-二亚硝基五亚甲基四胺的质量分数;ps为内标物的纯度;ah为n,n-二亚硝基五亚甲基四胺的峰面积;as为内标物的峰面积;nh为1摩尔n,n-二亚硝基五亚甲基四胺特征信号的官能团上共振核的数目;ns为1摩尔内标物特征信号的官能团上共振核的数目;mh为n,n-二亚硝基五亚甲基四胺的摩尔质量;ms为内标物的摩尔质量;mh为n,n-二亚硝基五亚甲基四胺的质量;ms为内标物的质量;(5)采用步骤(4)相同的操作测定并计算发泡剂中化合物三亚硝基三亚甲基三胺的质量分数。优选的,其中,步骤(3)中所述内标物选自对二氯苯或对二硝基苯,优选的,所述内标物为高纯品或标准品。优选的,其中,步骤(1)或步骤中(2)所用样品的质量为3~4g;优选地,步骤(3)中所用样品质量为30~60mg。优选的,其中,所述发泡剂产品中水分为0.20-0.29wt%,灰分为0.060-0.083wt%;优选地,三亚硝基三亚甲基三胺的含量为0.8-2.0wt%。优选的,其中,步骤(3)中氘代溶剂的用量为2.0-8.0ml;优选地,步骤(4)中内标物的质量为10-25mg。优选的,其中,步骤(4)中核磁共振氢谱测试条件包括:核磁波谱仪共振频率为400~600mhz;优选地,温度为20~35℃;优选地,脉冲角度为90°;优选地,延迟时间20~40s;优选地,采样次数为32~128次。优选的,其中,步骤(1)中进行干燥的温度为70~100℃,优选干燥时间为2~4小时。优选的,其中,步骤(2)中进行焙烧的温度为500~800℃,优选焙烧时间为2~4小时。优选的,其中,所述氘代溶剂为重氢二甲基亚砜(dmso-d6),氘代氯仿和氘代乙腈中的一种或两种以上;优选为重氢二甲基亚砜(dmso-d6)。本发明还提供了所述的方法在有机发泡剂纯度分析
技术领域:
的应用。本发明所述的发泡剂为含胺类的发泡剂。发泡剂h为主要含n,n-二亚硝基五亚甲基四胺的发泡剂。其中,所述n,n-二亚硝基五亚甲基四胺和三亚硝基三亚甲基三胺的摩尔质量为186.17和174.12g/mol,化学式为c5h10n6o2和c3h6n6o3,化学结构式为:所述n,n-二亚硝基五亚甲基四胺核磁共振氢谱特征峰为:synδ:5.23(d,j=13hz,2h),5.45(d,j=13hz,2h),4.22(d,j=14hz,2h),5.46(d,j=14hz,2h),4.46(s,1h),5.35(d,1h)。antiδ:5.35(d,j=13hz,2h),5.77(d,j=13hz,2h),4.29(d,j=14hz,2h),5.76(d,j=14hz,2h),4.46(s,2h)。所述三亚硝基三亚甲基三胺核磁共振氢谱特征峰:anti5.62(s,2h),6.26(s,2h),6.90(s,2h),syn6.22(s,2h),在本发明的一个优选实施方案中,提供了一种发泡剂h产品纯度的检测方法,其包括以下具体步骤:(1)在恒重的称量瓶中称取3~4g样品(准确至0.0001g),放置恒温干燥箱中干燥,自然冷却,计算样品中水分w%。(2)在恒重的坩埚中称取3~4g样品(准确至0.0001g),放置马弗炉中焙烧,自然冷却,计算样品中灰分s%。(3)称取30~60mgn,n-二亚硝基五亚甲基四胺和10~25mg内标物置于样品瓶中,加入2.0~8.0ml氘代溶剂溶解,对其进行核磁共振氢谱测试。(4)确定步骤(3)核磁共振氢谱中n,n-二亚硝基五亚甲基四胺和内标物的特征峰,测定其相对积分面积分别为ah和as,将数据代入下式得到测定n,n-二亚硝基五亚甲基四胺质量分数:其中ph为n,n-二亚硝基五亚甲基四胺的质量分数;ps为内标物的纯度;ah为n,n-二亚硝基五亚甲基四胺的峰面积;as为内标物的峰面积;nh为1摩尔n,n-二亚硝基五亚甲基四胺特征信号的官能团上共振核的数目;ns为1摩尔内标物特征信号的官能团上共振核的数目;mh为n,n-二亚硝基五亚甲基四胺的摩尔质量;ms为内标物的摩尔质量;mh为n,n-二亚硝基五亚甲基四胺的质量;ms为内标物的质量。采用相同方法测定三亚硝基三亚甲基三胺(发泡剂h副产物)的含量。在本发明的一个优选实施方案中,所述内标物为高纯品或标准品的对二氯苯、对二硝基苯。此两种内标物在核磁共振中达到的相应程度较好,而采用本领域常用的内标物,如对二苯酚为内标物,进行核磁共振测试时,苯环上氢的化学位移位于三亚硝基三亚甲基三胺特征峰中间,较为杂乱。在本发明的一个优选实施方案中,所述核磁共振氢谱测试条件:核磁波谱仪共振频率为400~600mhz,温度为20~35℃,脉冲角度为90o,延迟时间20~40s,采样次数为32~128次。在本发明的一个优选实施方案中,检测所用样品发泡剂h中副产物三亚硝基三亚甲基三胺(ttt)的含量为0.8-2.0wt%,水分为0.20-0.29wt%;灰分为0.060-0.083wt%。在本发明的一个优选实施方案中,所述核磁共振氢谱测试n,n-二亚硝基五亚甲基四胺纯度方法的准确率高达99%以上。为了验证用以检测n,n-二亚硝基五亚甲基四胺纯度的核磁共振氢谱法的最佳检测条件,进行了下述实施例的具体实验过程。下面对本发明作进一步详细描述。下面实施例中所用到各试剂和仪器来源如下表1:表1实施例所用试剂和仪器试剂/仪器纯度/型号厂家发泡剂h95.0-99.1%安徽润岳科技有限责任公司对二氯苯99.5%上海阿拉丁生化科技股份有限公司对二硝基苯99.9%上海阿拉丁生化科技股份有限公司氘代二甲亚砜dmso-d6国药集团化学试剂有限公司恒温干燥箱dhg-9070上海精宏实验设备有限核磁波谱仪avanceⅲ400或avanceneo600德国布鲁克公司实施例实施例1实施例1-1在恒重的称量瓶中称取3.1473g发泡剂h(样品1),放置80℃恒温干燥箱中,干燥4h,自然冷却,干燥后发泡剂h净重3.1381g,计算样品含水量为0.29%。在恒重的坩埚中称取3.6728g发泡剂h(样品1),放置马800℃弗炉中,焙烧2h,自然冷却,计算样品中灰分0.071%。称取40.5mg发泡剂h(样品1)和13.9mg对二氯苯(纯度99.5%)置于样品瓶中,加入2.00mldmso溶解,对其进行核磁共振氢谱测试,其核磁共振氢谱如图1所示,其中图中编号1、2和3的峰分别为n,n-二亚硝基五亚甲基四胺、三亚硝基三亚甲基三胺和内标物的峰。对核磁共振氢谱中n,n-二亚硝基五亚甲基四胺、对二氯苯(内标物)的特征峰进行积分,相对积分面积分别为ah1、as1,通过如下述的数学公式(1)经计算n,n-二亚硝基五亚甲基四胺含量为97.76%;其中,核磁波谱仪共振频率为400mhz,温度为20℃,脉冲角度为90°,延迟时间20s,采样次数为32次。然后采用相同的方法,根据所述核磁共振氢谱的图1-1上的三亚硝基三亚甲基三胺峰面积计算样品中三亚硝基三亚甲基三胺的含量为1.85%,实施例1的结果汇总于下面的表2中。实施例1-2实验过程与实施例1-1相同,仅将其中内标物对二氯苯(纯度99.5%)的质量修改为18.9mg。实施例1-3实验过程与实施例1-1相同,仅将其中内标物对二氯苯(纯度99.5%)的质量修改为24.8mg。实施例1-4实验过程与实施例1-1相同,仅将其中核磁波谱仪共振频率为600mhz,温度为35℃,脉冲角度为90°,延迟时间40s,采样次数为128次。实施例1-5实验过程与实施例1-1相同,仅将其中核磁波谱仪共振频率为400mhz,温度为28℃,脉冲角度为90°,延迟时间30s,采样次数为80次。表2实施例1发泡剂h产品纯度测试结果实施例2实施例2-1在恒重的称量瓶中称取3.4432g发泡剂h(样品2),放置90℃恒温干燥箱中,干燥3h,自然冷却,干燥后发泡剂h净重3.4363g,计算样品含水量为0.20%。在恒重的坩埚中称取3.1013g发泡剂h(样品2),放置660℃马弗炉中,焙烧3h,自然冷却,计算发泡剂h中灰分0.083%。称取41.8mg发泡剂h(样品2)和12.7mg对二硝基苯(纯度99.5%)置于样品瓶中,加入2.00mldmso溶解,对其进行核磁共振氢谱测试。对核磁共振氢谱中n,n-二亚硝基五亚甲基四胺、对二硝基苯的特征峰进行积分,相对积分面积分别为ah2、as2,经计算n,n-二亚硝基五亚甲基四胺含量为98.07%,其中,核磁波谱仪共振频率为400mhz,温度为30℃,脉冲角度为90°,延迟时间30s,采样次数为64次。然后采用相同的方法,根据所述核磁共振氢谱的图上的三亚硝基三亚甲基三胺峰面积计算样品中三亚硝基三亚甲基三胺的含量为1.49%,实施例2的结果汇总于下面的表3中。实施例2-2实验过程与实施例2-1相同,仅将其中内标物对二硝基苯(纯度99.5%)的质量修改为16.2mg。实施例2-3实验过程与实施例2-1相同,仅将其中内标物对二硝基苯(纯度99.5%)的质量修改为23.7mg。实施例2-4实验过程与实施例2-1相同,仅将其中核磁波谱仪共振频率为600mhz,温度为35℃,脉冲角度为90°,延迟时间40s,采样次数为128次。实施例2-5实验过程与实施例2-1相同,仅将其中核磁波谱仪共振频率为400mhz,温度为28℃,脉冲角度为90°,延迟时间30s,采样次数为80次。表3实施例2发泡剂h产品纯度测试结果实施例3准确度检测提前准备样品:将纯化合物n,n-二亚硝基五亚甲基四胺和三亚硝基三亚甲基三胺按照质量比99:1混合均匀,然后按照实施例1-1的检测方法进行检测,将检测得到的结果与原始混合含量进行对比,得到检测方法的准确度。用十万分之一天平称取682.26mgn,n-二亚硝基五亚甲基四胺(99.5%)、13.91mg三亚硝基三亚甲基三胺(纯度99.5%)和218.93mg对二氯苯(纯度99.5%)置于样品瓶中,加入10.00mldmso溶解,对其进行核磁共振氢谱测试。其中,核磁波谱仪共振频率为600mhz,温度为20℃,脉冲角度为90°,延迟时间20s,采样次数为32次。对磁共振氢谱中n,n-二亚硝基五亚甲基四胺、对二氯苯的特征峰进行积分,相对积分面积分别为ah3、at3和as3,经计算n,n-二亚硝基五亚甲基四胺和三亚硝基三亚甲基三胺含量分别为97.90%和1.97%;样品中n,n-二亚硝基五亚甲基四胺和三亚硝基三亚甲基三胺理论含量分别为98.00%和2.00%,实验结果准确率分别为99.89%和98.59%。对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1